
Accelerated Approval Under Scrutiny


The regulatory landscape of 2026 has witnessed a paradigm shift in the governance of therapeutic development for rare and ultra-rare conditions. For investors, biotechnology disruptors, and pharmaceutical leaders, the accelerated approval pathway has transitioned from a flexible, albeit somewhat unpredictable, mechanism into a highly formalized and rigorously overseen strategic instrument. This transformation, catalyzed by the Food and Drug Omnibus Reform Act of 2022 (FDORA) and subsequent operationalizations by the U.S. Food and Drug Administration (FDA) through April 2026, fundamentally alters the risk profile for drug development. At the center of this shift is the agency's evolving stance on surrogate endpoints those laboratory measurements, radiographic images, or physical signs that are not themselves measures of clinical benefit but are "reasonably likely" to predict such benefit.
The current environment is defined by a dual-pronged regulatory philosophy. On one hand, the FDA is exercising unprecedented flexibility for ultra-rare conditions through frameworks like the "Plausible Mechanism Pathway," which allows for marketing authorization based on biological plausibility and target engagement when traditional randomized controlled trials (RCTs) are infeasible. On the other hand, the agency is enforcing much stricter requirements for post-marketing confirmatory trials, demanding that these studies be "underway" at the time of approval and granting itself expedited powers to withdraw drugs from the market if clinical benefit is not verified. This report provides an exhaustive analysis of these shifts, the statistical performance of the pathway in 2025, and the strategic implications for the life sciences investment community.
The Legislative Evolution: From 1992 to FDORA 2022
The Accelerated Approval Program was born out of the HIV/AIDS crisis in 1992, established via rulemaking to expedite access to life-saving therapies for serious conditions with unmet medical needs. While the program was later codified under the Food and Drug Administration Safety and Innovation Act of 2012 (FDASIA), it remained subject to criticism regarding the "dangling" status of many approvals indications that remained on the market for years without ever completing the required confirmatory studies. A landmark 2022 report from the Office of Inspector General (OIG) underscored these concerns, revealing that 34% of incomplete confirmatory trials were past their original planned completion dates, some by more than a decade.
In response, the U.S. Congress enacted FDORA as part of the Consolidated Appropriations Act of 2023. Section 3210 of FDORA granted the FDA significant new authorities to ensure the integrity of the pathway. These authorities, which were fully integrated into agency operations by 2025 and early 2026, include the power to require that confirmatory trials be "underway" prior to approval and the creation of an expedited withdrawal process that bypasses the lengthy public hearings of the past.
Table 1: Institutional Oversight and AACC Membership (2025-2026)
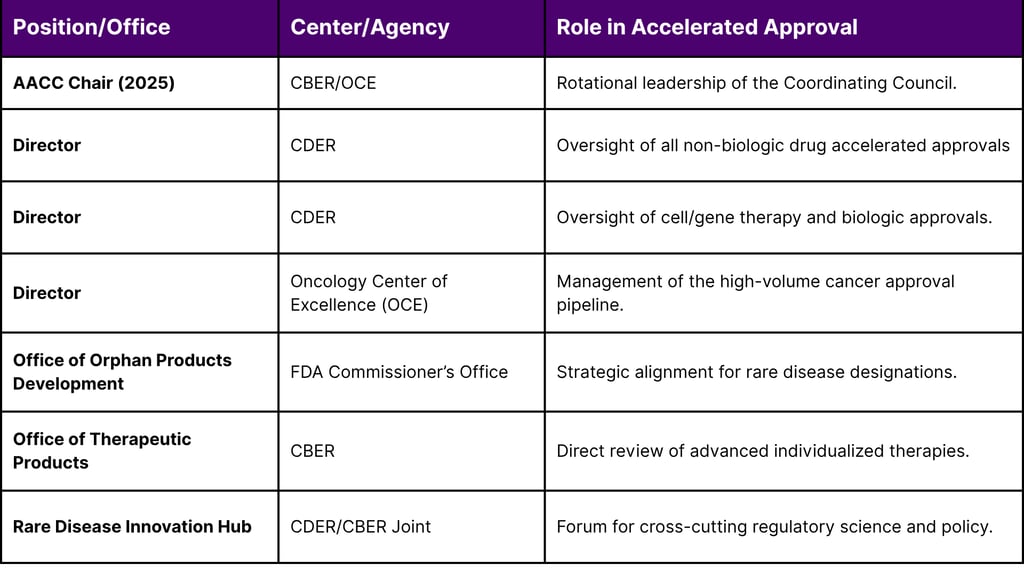
The Burden of Being "Underway": Redefining Post-Approval Commitment
One of the most significant changes for investors to navigate is the FDA's new interpretation of what constitutes a trial being "underway." Historically, drug sponsors could wait until after they had secured marketing authorization and the associated cash flow to begin the difficult work of enrolling patients in confirmatory studies. This led to a conflict of interest: once a drug was available on the market, patients were often unwilling to enroll in a randomized trial where they might receive a placebo or an inferior control.
Under the draft guidance finalized in early 2026, the FDA clarifies that "underway" typically means that enrollment has been initiated or that significant progress toward site activation has been achieved. For investors, this front-loads the capital expenditure of a drug program. A sponsor can no longer defer the costs of a Phase 3-style confirmatory study until after commercial launch. Instead, the "registration-defining" trial must often be in progress simultaneously with the primary review period.
Table 2: Confirmatory Trial Compliance and Regulatory Actions (2022-2025)
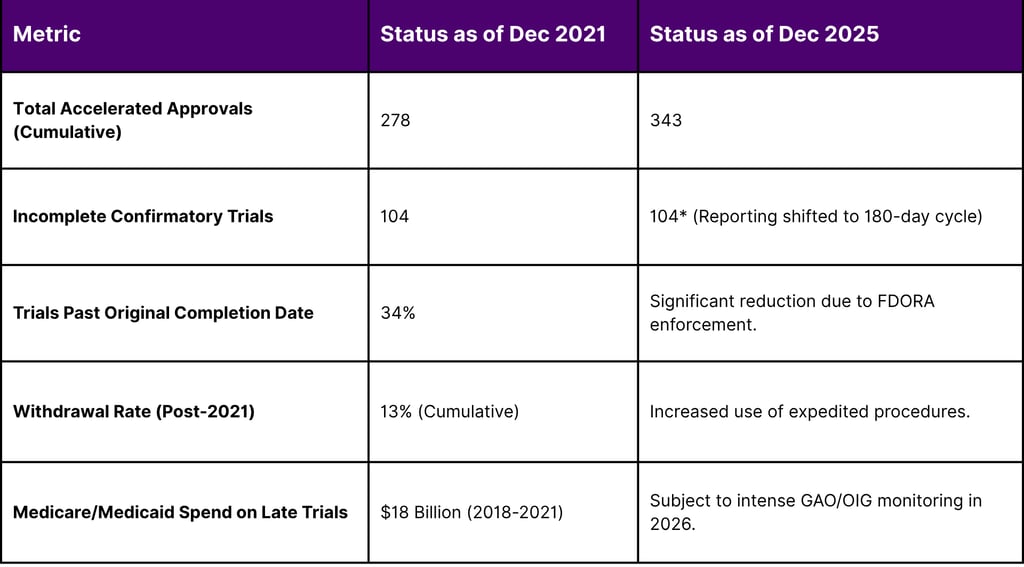
The Plausible Mechanism Framework: A Pivot Toward Biological Integrity
While the FDA has tightened the rules for common conditions, it has opened a new door for ultra-rare diseases. On February 23, 2026, the agency issued the "Plausible Mechanism Framework" Draft Guidance, a document that represents a revolutionary advance in regulatory science. This framework specifically addresses individualized therapies, such as genome editing (GE) and RNA-based therapies like antisense oligonucleotides (ASOs), intended for diseases where randomized trials are simply not feasible due to extremely small patient populations.
The framework shifts the evidentiary burden from broad clinical outcomes to the "how" of the disease. If a sponsor can identify a specific genetic, cellular, or molecular abnormality and demonstrate with high precision that the therapy targets that root cause, the FDA may grant approval based on target engagement and a "highly supported plausible mechanism of action". This is particularly relevant for "n-of-1" therapies where only one or a few patients exist worldwide.
For the investment community, this framework creates a path to value for "platform-based" companies. Under the guidance, a sponsor using CRISPR or ASO technology can submit a single application covering multiple targets within a single gene if the method of correction is consistent. This "one-application, many-mutations" approach dramatically reduces the regulatory hurdle for scaling bespoke treatments. However, the agency compensates for this flexibility by imposing heavy post-marketing commitments, often requiring de-identified real-world data and natural history cohorts to supplement the small safety datasets.
Statistical Insights: The 2025 Approval Landscape
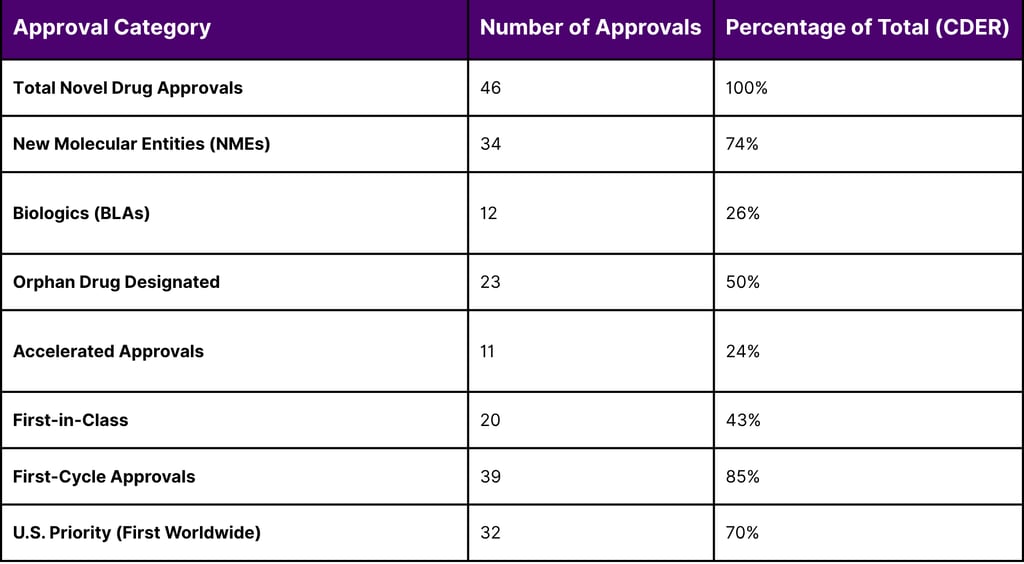
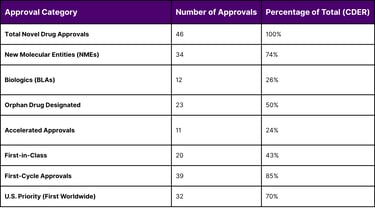
The year 2025 was a benchmark year for pharmaceutical innovation, with the FDA maintaining a high volume of approvals despite significant internal leadership transitions and the implementation of new oversight councils. The Center for Drug Evaluation and Research (CDER) approved 46 novel drugs, while the Center for Biologics Evaluation and Research (CBER) contributed 12, for a total of 58 novel approvals across the agency.
A granular analysis of these approvals reveals the dominance of the rare disease sector. Exactly 50% (23 out of 46) of CDER's novel approvals received Orphan Drug Designation, intended for diseases affecting fewer than 200,000 people in the U.S. Furthermore, 24% of these novel drugs utilized the accelerated approval pathway, emphasizing that despite the "scrutiny" mentioned in recent guidelines, the pathway remains a critical engine for the biotech economy.
Table 3: CDER Novel Drug Approvals and Regulatory Metrics (Full Year 2025)
The efficiency of the review process remains high, with the FDA meeting or exceeding its Prescription Drug User Fee Act (PDUFA) goal dates for 96% of novel approvals in 2025. However, the "quality over quantity" shift is evident in the 19 complete response letters (CRLs) issued for 18 novel drugs that failed to meet statutory requirements. Since September 2025, the FDA has begun making these CRLs public, providing investors with unprecedented insight into the specific reasons for drug rejections ranging from failed surrogate endpoint validation to manufacturing deficiencies.
Surrogate Endpoints and the "One Pivotal Trial" Default
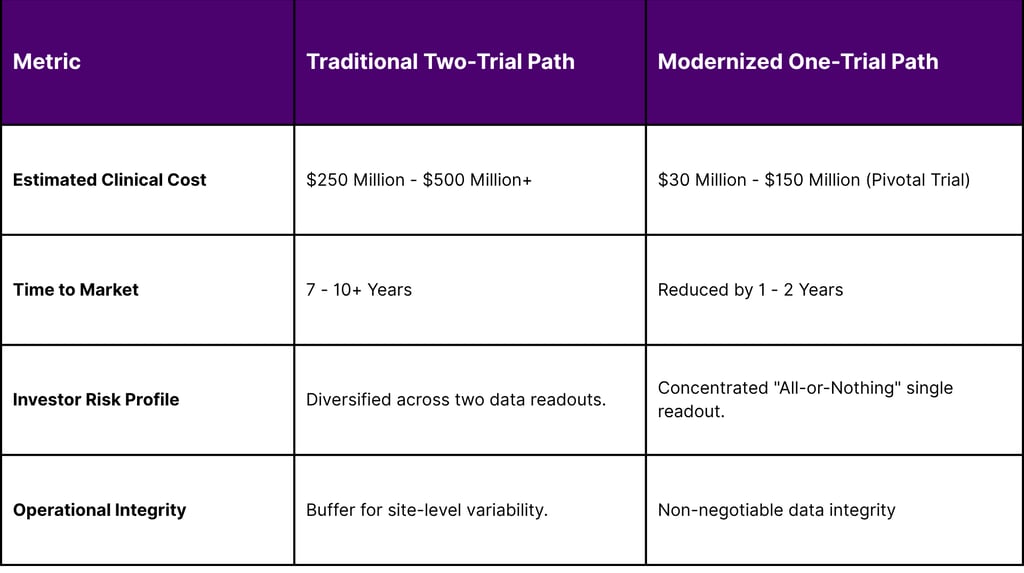
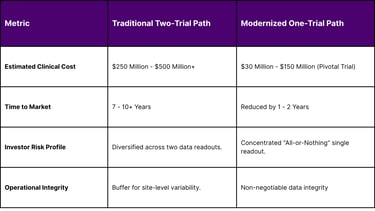
In a move that has profound implications for trial design and R&D budgets, the FDA signaled a shift in February 2026 away from the "two-trial dogma." For decades, the gold standard for "substantial evidence of effectiveness" was two independent, well-controlled studies. As of 2026, the agency has publicly positioned a single robust pivotal trial, supported by confirmatory evidence, as the new general default approach for drug approval.
This "one-trial" approach is not a relaxation of standards but a shift toward "evidence optimization." The agency now emphasizes the "totality-of-evidence narrative," where a single pivotal trial is bolstered by mechanistic and translational data, biomarker alignment, and prior clinical experience.
Table 4: Economic Impact of Trial Reductions and Delays
The Rare Disease Endpoint Advancement (RDEA) Pilot Program
To facilitate the development of novel surrogate endpoints, the FDA established the RDEA Pilot Program under PDUFA VII and FDORA. Rare diseases often lack validated biomarkers or clinical outcome assessments (COAs), making it difficult for sponsors to design trials that satisfy traditional approval standards. The RDEA program offers sponsors up to three follow-up meetings with the FDA to collaborate on endpoint development.
As of April 2026, the program is actively reviewing proposals on a quarterly basis, with a maximum of three accepted per year. A notable example is Abeona Therapeutics, which had its ABO-503 gene therapy for X-linked retinoschisis selected for the program. This collaboration allows for "frequent advice and regular ad-hoc conversations" to validate novel efficacy markers, significantly reducing the regulatory risk for the program's investors.
The agency's commitment to this area is further evidenced by the "Advancing Novel Surrogate Endpoints For Rare Disease Drug Development Workshop" scheduled for May 18, 2026. This public forum will discuss digital health technologies and the use of de-identified patient-level real-world data (RWD) to support novel surrogates in marketing applications.
Rare Disease Approvals: A Detailed Look at Q1 2026
The first quarter of 2026 has already yielded several landmark approvals for rare conditions, illustrating the practical application of the FDA's flexible review mechanisms. Many of these therapies are the first ever approved for their respective indications, addressing profound unmet needs.
Table 5: Notable Rare Disease Drug Approvals (Q4 2025 - Q1 2026)
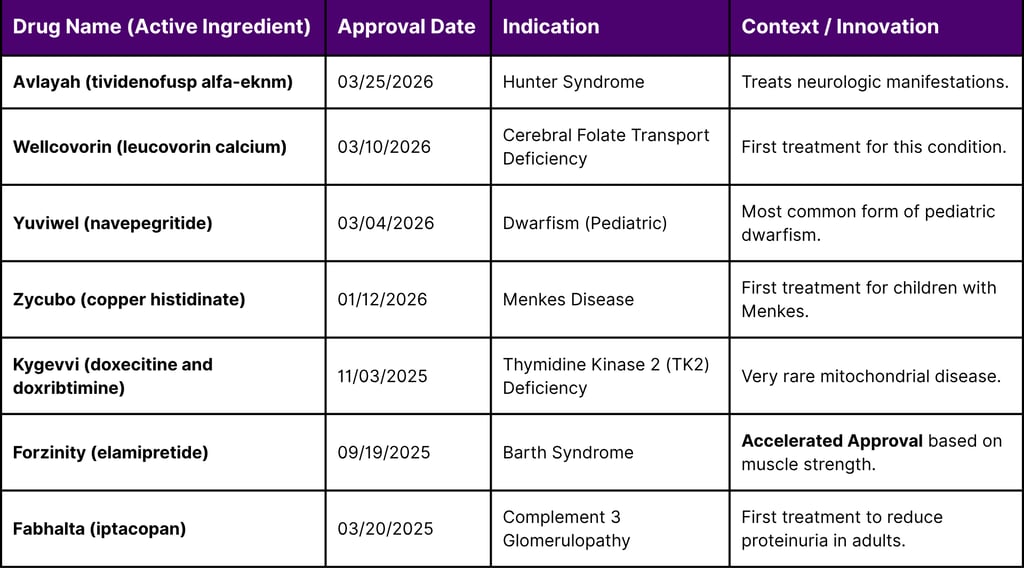
The approval of Forzinity (elamipretide) for Barth Syndrome in late 2025 provides a high-profile case study of the new "surrogate endpoint" rigor. Barth Syndrome is an infantile-onset mitochondrial disease with no previous approved treatments. The FDA concluded that while the primary endpoint (knee extensor muscle strength) was only an intermediate clinical measure and not a direct clinical benefit, it was "reasonably likely" to predict improvement in physical functioning, thus justifying accelerated approval while a confirmatory trial verifies clinical benefit.
The Rare Disease Innovation Hub and Strategic Agenda 2026
To provide a single point of connection for the rare disease community, the FDA launched the Rare Disease Innovation Hub in late 2025.14 This cross-center program serves as a forum for CDER and CBER to collaborate on cross-cutting issues like "n-of-1" trial designs and the identification of innovative biomarkers.
On February 2, 2026, the Hub released its Strategic Agenda, which outlines three primary goals for the year:
Regulatory Science Advancement: Creating standardized "Rare Disease Evidence Principles" (RDEP) to clarify what kinds of data including non-clinical models and expanded access data can support a finding of "substantial evidence".16
Coordination and Alignment: Ensuring that when a disease population is small, both drug and biologic reviewers apply the same levels of regulatory flexibility.
Centralized Connection: Consolidating website resources and launching a quarterly newsletter to keep the community informed of noteworthy approvals and program updates.
The Hub's creation reflects an institutional admission that rare disease drug development is fundamentally different from mass-market pharmaceuticals. For investors, the Hub represents a reduction in "procedural friction," offering a more predictable pathway for therapies targeting small patient populations.
The Risks of Success: Pricing and "March-In" Rights
The economic viability of rare disease pipelines is increasingly tied to the political and regulatory debate over drug pricing. In 2025 and 2026, the Government Accountability Office (GAO) and other federal bodies have heightened their focus on the high cost of accelerated approval drugs. Because these drugs enter the market before their clinical benefit is fully confirmed, they are often the target of "march-in" rights discussions.
A February 2026 GAO report examined the potential impacts of using product price as a factor for exercising march-in rights under the Bayh-Dole Act. While march-in rights have never been exercised by a federal agency, the 2023 draft guidance from the National Institute of Standards and Technology (NIST) proposed that high prices could be a criterion for the government to step in and license a federally funded invention to other parties. Although studies suggest this would likely affect only a small number of drugs (as most do not have patents subject to Bayh-Dole), the mere possibility of price-based march-in creates a new layer of terminal-value risk for investors.
Furthermore, the 2022 OIG report highlighted that Medicare and Medicaid spent over $18 billion on accelerated approval drugs whose confirmatory trials were late. This has led to proposals for dynamic pricing models where a drug's price is "rebated" or reduced if confirmatory milestones are missed.
Withdrawal Procedures and "Dangling" Indications
The "dangling" approval where a drug remains on the market despite a failed confirmatory trial is a target of the new expedited withdrawal procedures. Historically, the FDA relied on voluntary withdrawals from sponsors, a process that was "slow and often ineffective". High-profile examples like Aducanumab (Aduhelm), which was withdrawn in late 2024 after failing to verify benefit, highlight the new regulatory pressure.
The new process mandated by FDORA gives the FDA a procedure for withdrawal that does not involve a public hearing and is intended to take far less time. The sponsor still has the opportunity for a meeting and a written appeal to the Commissioner, but the agency's ability to act decisively has been significantly strengthened.
Investors must now factor in "post-approval failure" as a high-probability event if trial designs are not robust. In 2024 and 2025, several indications for major oncology drugs were voluntarily withdrawn after negative post-approval study results, reflecting a new industry-wide trend toward compliance with the "Project Confirm" initiative.
Future Outlook: Modernizing Biotechnology Regulation
As of April 2026, the FDA is proposing further legislative reforms to modernize clinical trials for biotechnology products. The National Security Commission on Emerging Biotechnology (NSCEB) has published recommendations to streamline regulatory review to keep the U.S. competitive with other global biopharmaceutical powers. These proposals include validating more modern testing methods such as "New Approach Methodologies" (NAMs) to reduce the reliance on animal testing and accelerate the pre-clinical phase.
The agency's "Rare Disease Day 2026" event on February 23 emphasized the dual importance of the "patient voice" and "real-world evidence". As the Hub begins to fund dedicated staff and quarterly newsletters, the flow of information between regulators and developers is expected to reach an all-time high.
FAQ
What constitutes a trial being "underway" in 2026?
According to 2025-2026 draft guidance, "underway" typically requires that enrollment has been initiated or significant progress has been made toward site activation by the date of approval.
How does the Plausible Mechanism Framework differ from traditional accelerated approval?
While traditional accelerated approval uses surrogate endpoints reasonably likely to predict clinical benefit, the Plausible Mechanism Framework allows for approval of individualized therapies for ultra-rare diseases based on biological target engagement and mechanistic rationale when RCTs are not feasible.
What are "march-in" rights, and how do they affect drug pricing?
March-in rights allow the government to license a federally funded invention to other parties under specific criteria. A 2026 GAO report discussed using "high price" as a factor for exercising these rights, though they have never been used to date.
What was the "Orphan Drug" share of approvals in 2025?
Exactly 50% (23 out of 46) of the novel drugs approved by CDER in 2025 received Orphan Drug Designation, emphasizing the sector's importance to the pharmaceutical pipeline.
Can the FDA withdraw an accelerated approval drug without a public hearing?
Yes. Under FDORA authorities implemented in 2024-2025, the FDA now has an expedited withdrawal process that bypasses the lengthy public hearing requirement of the past.
References
A Quick Take on FDA's New Draft Accelerated Approval Guidance for Drug Products
Accelerated Approval Council Activities Report CY 2025 ... - FDA
Accelerated Approvals | FDA
Accelerated Approval Program - FDA
FDA Advances a “Plausible Mechanism” Framework for Rare Disease Drug Development and Signals a Shift to a Single Trial with Confirmatory Evidence Default for All Drugs | Advisories | Arnold & Porter
FDA Launches Framework for Accelerating Development of Individualized Therapies for Ultra-Rare Diseases
Accelerated Approval and Considerations for Determining Whether a Confirmatory Trial is Underway January 2025 - FDA
Three Overhauls to Accelerated Drug Approval You Need to Know About: Novel Endpoints, Confirmatory Trials, and Expedited Withdrawal of Approvals | ArentFox Schiff
Rethinking FDA's Accelerated Approval Pathway: New Draft Guidances and Implications for Drug Companies - McGuireWoods
Project Confirm | FDA
Accelerated Approval confirmatory trials requirement may further complicate life science deals
FDA's Latest Thinking on Accelerated Approval Program for Drugs and Biologics - Dentons Lee
Delays in Confirmatory Trials for Drug Applications Granted FDA's Accelerated Approval Raise Concerns | Office of Inspector General
FDA Rare Disease Innovation Hub
FDA Rare Disease Innovation Hub Q&A
Rare Disease Month Developments, Part 1 – The Good: RPD PRV Program Renewed, FDA Rare Disease Hub's 2026 Strategic Agenda & Plausible Mechanism Draft Guidance On Its Way
Public Comment of National Center for Health Research Regarding Accelerated Approval and Considerations for Determining Whether a Confirmatory Trial is Underway
Accelerated Approval and Considerations for Determining Whether a Confirmatory Trial is Underway; Draft Guidance for Industry; Availability - Federal Register
FDA's Shift to One Pivotal Trial for Drug Approval: Regulatory Strategy Implications for Sponsors - ProPharma
The FDA's Shift to One Pivotal Trial: What It Means for Modern Development Programs
Regulatory NewsBREAK: The FDA Releases Draft Framework to Accelerate Individualized Therapies for UltraRare Diseases | AMCP.org
WTAS: FDA Launches Framework for Accelerating Development of Individualized Therapies for Ultra-Rare Diseases | HHS.gov
Rare disease round-up: FDA efforts to advance treatments - Hogan Lovells
US FDA Holds Steady With 58 Novel Approvals In 2025 Despite Upheaval
FDA CDER Releases 15th Annual New Drug Therapy Approvals Report for 2025
Advancing Health Through Innovation: New Drug Therapy ... - FDA
Accelerated Approvals Perked Up While US FDA Excelled At Being Average In 2025
FDA Legislative Proposals Would Modernize and Streamline Regulatory Process for Promising Medical Products - National Security Commission on Emerging Biotechnology
FDA: One pivotal trial (+ confirmatory evidence) is default expectation for approval - AMR.Solutions
Advancing Novel Surrogate Endpoints For Rare Disease Drug Development Workshop - 05/18/2026 | FDA
Advancing Novel Surrogate Endpoints For Rare Disease Drug Development Workshop - 05/18/2026 | FDA
Learning and Education to ADvance and Empower Rare Disease Drug Developers (LEADER 3D) | FDA
Submission Deadlines and Process | Rare Disease Endpoint Advancement Pilot Program
Abeona Therapeutics' ABO-503 chosen for FDA's Rare Disease Endpoint Advancement Pilot Program | Ophthalmology Times - Clinical Insights for Eye Specialists
Rare Disease Drug Approvals - FDA
Press Announcements | FDA
215244Orig1s000 INTEGRATED REVIEW - accessdata.fda.gov
CDER/CBER Rare Disease Evidence Principles (RDEP) - FDA
Intellectual Property: Information on Draft Guidance to Assert Government Rights Based on Price | U.S. GAO
Strengthening the FDA's Accelerated Approval Pathway: Progress and Unfinished Business
Industry promotion of oncology drugs with accelerated approval that failed confirmatory trials. - ASCO Publications
Navigation
© 2026 FyreIgnis Market Research. All rights reserved.
Legal
info@fyreignismarketresearch.com
India
