
Beyond CRISPR: The Regulatory Pathway of Base and Prime Editing Technologies


The gene editing landscape has evolved dramatically since December 2023, when the FDA approved Casgevy (exagamglogene autotemcel), marking the first CRISPR/Cas9-based therapy authorization in the United States. This landmark approval for sickle cell disease treatment signaled a new era for precision medicine, yet it also highlighted the ongoing evolution toward even more refined editing technologies: base editing and prime editing.
This comprehensive analysis examines the regulatory pathway and clinical progression of next-generation gene editing technologies from 2024 through early 2026, with projections for regulatory milestones expected in 2026-2027. Drawing exclusively from government sources including the FDA, NIH, and ClinicalTrials.gov, this report provides an evidence-based assessment of how base editing and prime editing are advancing from laboratory innovation to clinical application.
Introduction
Gene editing has transitioned from theoretical possibility to clinical reality with remarkable speed. The December 2023 FDA approval of Casgevy, developed by Vertex Pharmaceuticals and CRISPR Therapeutics, demonstrated that CRISPR-based therapies could meet rigorous safety and efficacy standards. Casgevy utilizes CRISPR/Cas9 technology to edit hematopoietic stem cells, targeting the BCL11A gene to induce fetal hemoglobin production in patients with sickle cell disease and transfusion-dependent beta thalassemia.
However, traditional CRISPR-Cas9 technology creates double-strand breaks in DNA, which can result in unintended genetic consequences through imprecise cellular repair mechanisms. This limitation has driven development of more precise alternatives: base editing and prime editing. These technologies promise to rewrite genetic information without generating double-strand DNA breaks, potentially offering enhanced precision and reduced off-target effects.
Understanding Next-Generation Gene Editing Technologies
Base Editing: Single-Letter Precision
Base editing technology, first published in 2016, enables direct conversion of one DNA base pair into another without creating double-strand breaks. Base editors fuse a catalytically impaired Cas enzyme to a deaminase enzyme, producing highly predictable single-nucleotide conversions:
Cytosine base editors (CBEs) convert C•G base pairs to T•A
Adenine base editors (ABEs) convert A•T base pairs to G•C
Glycosylase base editors (GBEs) enable C-to-G conversions
Dual-base editors (DBEs) perform simultaneous conversions
According to FDA guidance on genome editing products, base editors are considered gene therapy products that 'mediate their effects by specifically altering host genetic sequences.' The agency classifies these as human gene therapy products subject to Investigational New Drug (IND) application requirements under 21 CFR 312.23.
Prime Editing: Comprehensive DNA Rewriting
Prime editing, introduced in 2019, represents an evolution beyond base editing by enabling insertions, deletions, and all 12 possible base-to-base conversions without requiring double-strand breaks or donor DNA templates. The technology uses a prime editing guide RNA (pegRNA) that both directs the editor to the target site and provides the template for the desired edit.
In April 2024, the FDA cleared the first prime editing clinical trial (PM359) designed to treat chronic granulomatous disease. This milestone marked prime editing's transition from preclinical research to human clinical investigation, occurring approximately five years after the technology's initial publication.
FDA Regulatory Framework for Gene Editing Products
Classification and Oversight
The FDA regulates gene editing products through its Center for Biologics Evaluation and Research (CBER). According to FDA guidance on 'Human Gene Therapy Products Incorporating Human Genome Editing,' these products fall under the definition of biological products in section 351(i) of the Public Health Service Act.
The FDA's regulatory approach emphasizes several key principles:
Product-specific risk assessment based on mechanism of action
Evaluation of both on-target and off-target editing effects
Assessment of delivery vehicle safety and efficacy
Long-term follow-up monitoring requirements
Potential for germline transmission evaluation
The FDA has issued specific guidance on manufacturing, testing, and clinical trial design for genome editing products. These guidelines require sponsors to provide comprehensive data on product design, manufacturing processes, characterization, and preclinical safety assessment before initiating clinical trials.
Expedited Development Pathways
Several regulatory designations have accelerated gene editing therapy development:
Regenerative Medicine Advanced Therapy (RMAT) designation provides intensive FDA guidance and potential priority review
Fast Track designation enables more frequent FDA interactions and rolling review
Orphan Drug designation provides development incentives for rare diseases
Rare Pediatric Disease designation can grant a priority review voucher
Casgevy received all four designations during its development, demonstrating how these pathways can expedite regulatory review for transformative therapies addressing serious unmet medical needs.
Current Clinical Trial Landscape
Base Editing Clinical Programs
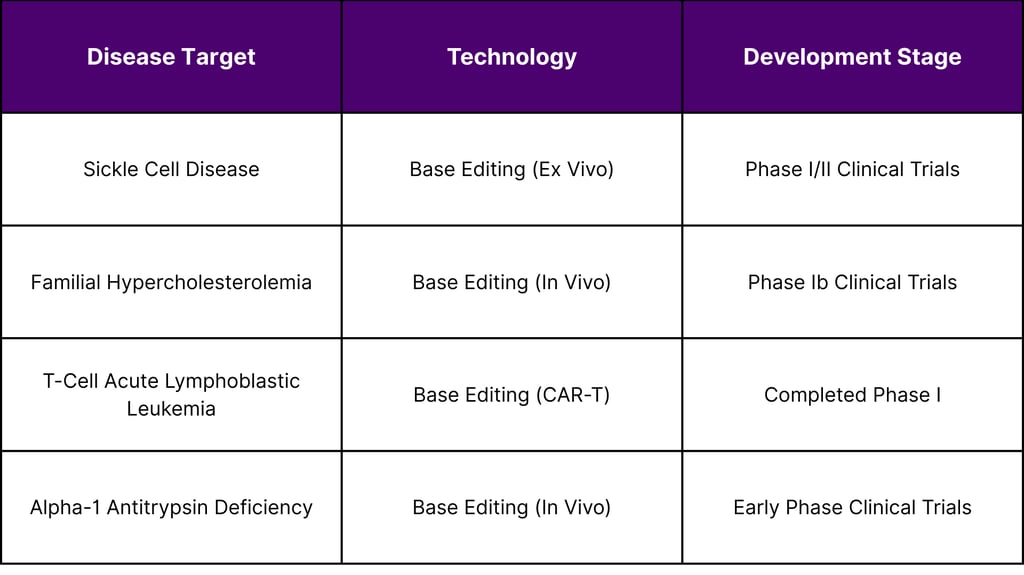
As of February 2026, base editing technologies have entered clinical investigation for multiple indications. While comprehensive trial data remains limited in publicly available government databases, several programs have reached human testing:
Prime Editing Clinical Development
Prime editing entered clinical trials in 2024, representing a faster bench-to-bedside translation than base editing. The first FDA-cleared prime editing trial (PM359) targets chronic granulomatous disease, an inherited immune disorder. This ex vivo approach involves editing patient hematopoietic stem cells outside the body before reinfusion.
In May 2025, researchers reported treating the first patient with a customized base editing therapy for a rare metabolic disorder, demonstrating the technology's potential for personalized medicine applications. The treatment was delivered in vivo via adeno-associated virus (AAV) vectors targeting liver cells.
Projected Regulatory Milestones: 2026-2027
Near-Term Approval Prospects
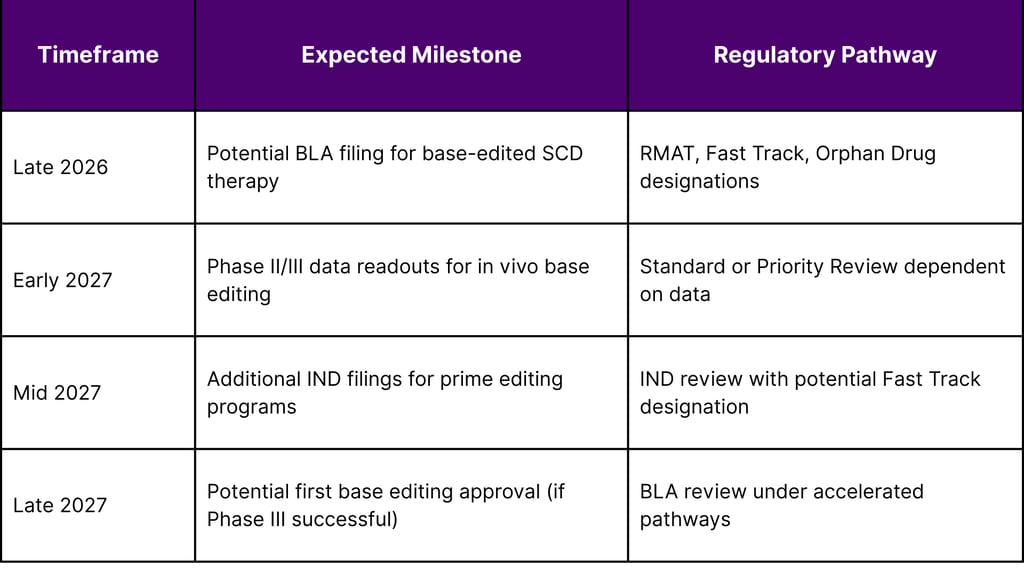
Based on current clinical trial progression and FDA guidance timelines, several base editing therapies could reach regulatory filing stages in 2026-2027:
Factors Influencing Approval Timelines
Several factors will determine whether these technologies achieve regulatory approval in the 2026-2027 timeframe:
Long-term safety data demonstrating absence of significant off-target effects
Durable efficacy in addressing disease pathology
Manufacturing consistency and product characterization
Delivery vehicle safety and tissue-specific targeting capabilities
Clinical trial enrollment and completion rates
The FDA's experience with Casgevy provides a regulatory precedent that may expedite review of subsequent gene editing therapies, particularly those demonstrating comparable or superior safety profiles.
Safety and Efficacy Considerations
Off-Target Effects and Monitoring
While base editing and prime editing avoid double-strand breaks, they are not without potential risks. FDA guidance emphasizes comprehensive assessment of:
Cas9-independent off-target editing at genomically similar sequences
Bystander editing of adjacent bases within the editing window
Delivery vehicle-related immune responses
Potential for transient mismatch repair pathway disruption
Recent research has identified that some base editors can produce unintended mutations independent of Cas9 targeting. These findings have prompted development of improved editor variants with reduced off-target activity while maintaining on-target efficiency.
In 2025, one NIH-led trial of a base-edited cell therapy was temporarily paused after a patient experienced a severe adverse event, underscoring that gene editing technologies require rigorous safety monitoring even without DNA double-strand breaks.
Delivery Challenges for In Vivo Applications
While ex vivo gene editing (editing cells outside the body) has progressed rapidly to clinical application, in vivo delivery presents additional challenges:
Adeno-associated virus (AAV) vectors have packaging size limitations (~4.7 kb) that constrain delivery of larger editing components
Lipid nanoparticles can trigger immune responses and have variable tissue targeting
Achieving therapeutic editing levels in target tissues while minimizing off-target tissue exposure
Managing pre-existing immunity to viral vectors in patient populations
Researchers are developing dual AAV systems, engineered virus-like particles, and non-viral delivery methods to address these limitations. FDA guidance requires thorough characterization of delivery vehicles and assessment of their contribution to overall product safety profile.
Market Access and Clinical Implementation
Pricing and Reimbursement Landscape
Casgevy's $2.2 million price point has established a reference for gene editing therapy costs. This pricing reflects:
Complex manufacturing processes requiring individualized cell collection and editing
Specialized treatment center infrastructure and expertise
Long-term patient monitoring and follow-up requirements
Development costs for innovative biotechnology
The Centers for Medicare & Medicaid Services (CMS) has implemented the Cell and Gene Therapy Access Model to facilitate broader access through outcomes-based agreements. Casgevy has secured Medicare add-on payments and Medicaid coverage in states with high sickle cell disease prevalence.
Treatment Center Infrastructure
As of mid-2025, 75 authorized treatment centers worldwide have been activated for Casgevy administration. These centers require:
Expertise in hematopoietic stem cell transplantation
Capacity for cell collection (apheresis)
Facilities for conditioning chemotherapy administration
Long-term follow-up infrastructure for safety monitoring
Base editing and prime editing therapies will require similar infrastructure, potentially limiting initial geographic access to major academic medical centers and specialized treatment facilities.
Future Directions and Emerging Trends
Disease-Agnostic Approaches
Recent research published in Nature (2025) demonstrates prime editing's potential for disease-agnostic treatment strategies. Investigators successfully used prime editing to convert endogenous tRNA genes into suppressor tRNAs, enabling readthrough of premature stop codons that cause approximately 11% of genetic diseases.
This approach could theoretically treat multiple diseases caused by nonsense mutations with a single therapeutic strategy, potentially accelerating development timelines and reducing costs through shared regulatory pathways.
Personalized N-of-1 Therapies
The successful treatment of a patient with a customized base editing therapy in May 2025 demonstrated proof-of-concept for individualized genetic medicines. Researchers completed diagnosis, editor optimization, toxicity studies, and FDA approval in under seven months.
This model could enable treatment of ultra-rare genetic diseases affecting only a few patients worldwide. However, it requires new regulatory frameworks for proportionate oversight based on benefit-risk assessment with limited clinical trial data. Proposed Centers for Interventional Genetics could streamline this process through standardized platforms and accelerated regulatory pathways.
Expansion Beyond Monogenic Diseases
While initial gene editing applications focus on single-gene disorders, researchers are exploring applications in:
Cardiovascular disease through permanent lipid modification
Neurodegenerative disorders via in vivo central nervous system delivery
Complex polygenic conditions through multi-target editing strategies
Infectious diseases including HIV through receptor modification
These applications will require advancement in delivery technology, multiplexed editing capabilities, and comprehensive long-term safety assessment.
Conclusion
Base editing and prime editing represent the next generation of precision genetic medicine, offering potential advantages over first-generation CRISPR-Cas9 approaches through elimination of double-strand DNA breaks. The regulatory pathway established by Casgevy's approval provides a framework for evaluating these technologies, while FDA guidance on genome editing products offers clear expectations for safety and efficacy demonstration.
Clinical trial progression suggests that base editing therapies for sickle cell disease and familial hypercholesterolemia could reach regulatory filing stages in late 2026 or early 2027, with potential approvals following in 2027-2028 if Phase III data demonstrate safety and efficacy. Prime editing remains earlier in clinical development, with initial trials focusing on chronic granulomatous disease and rare metabolic disorders.
The coming 12-18 months will be critical in determining whether these technologies deliver on their promise of enhanced precision and safety. Key milestones to monitor include Phase II/III trial results, additional IND filings, FDA guidance updates, and infrastructure development for commercial delivery.
As the field advances from treating rare monogenic disorders toward more common complex diseases, regulatory frameworks may need to evolve to accommodate personalized therapies, combination editing strategies, and novel delivery platforms. The FDA's adaptive approach to gene editing regulation will be essential in balancing innovation with patient safety as these transformative technologies move toward widespread clinical application.
Frequently Asked Questions
Q: How do base editing and prime editing differ from traditional CRISPR-Cas9?
A: Traditional CRISPR-Cas9 creates double-strand breaks in DNA, relying on cellular repair mechanisms that can produce unpredictable results. Base editing converts individual DNA bases without breaks, while prime editing can make insertions, deletions, and all base conversions using a template-guided mechanism, also without double-strand breaks.
Q: When will base editing or prime editing therapies be available to patients?
A: Based on current clinical trial timelines and regulatory pathways, the first base editing therapy could potentially receive FDA approval in late 2027 or 2028, pending successful Phase III trial completion and regulatory review. Prime editing therapies remain earlier in development and are unlikely to reach approval before 2028-2029.
Q: Are base editing and prime editing safer than traditional CRISPR?
A: These technologies avoid double-strand DNA breaks, which theoretically reduces certain risks associated with imprecise repair. However, they have their own safety considerations including Cas9-independent off-target editing and potential immune responses to delivery vehicles. Long-term clinical data will be necessary to fully assess their safety profiles.
Q: What diseases are being targeted by base editing and prime editing?
A: Current clinical programs focus on sickle cell disease, familial hypercholesterolemia, chronic granulomatous disease, alpha-1 antitrypsin deficiency, T-cell leukemia, and various rare metabolic disorders. Research is expanding into neurodegenerative diseases, cardiovascular conditions, and infectious diseases.
Q: How much will these therapies cost?
A: Pricing for base editing and prime editing therapies is expected to be comparable to Casgevy's $2.2 million price point for one-time treatments. In vivo therapies delivered via viral vectors may have different cost structures. Reimbursement models are evolving to address high upfront costs through outcomes-based agreements.
Q: What regulatory designations can accelerate development?
A: The FDA offers Regenerative Medicine Advanced Therapy (RMAT), Fast Track, Orphan Drug, and Rare Pediatric Disease designations that can expedite development, provide more frequent FDA interactions, enable rolling reviews, and potentially shorten approval timelines for therapies addressing serious unmet medical needs.
References
Centers for Medicare & Medicaid Services. (2025). Cell and gene therapy access model. U.S. Department of Health and Human Services.
ClinicalTrials.gov. (2024-2026). Clinical trials database [Data set]. U.S. National Library of Medicine.
U.S. Food and Drug Administration. (2023, December 8). FDA approves first gene therapies to treat patients with sickle cell disease [Press release].
U.S. Food and Drug Administration. (2024). Guidance for industry: Foods derived from plants produced using genome editing. Center for Food Safety and Applied Nutrition.
U.S. Food and Drug Administration. (2024). Guidance for industry: Human gene therapy products incorporating human genome editing. Center for Biologics Evaluation and Research.
U.S. Food and Drug Administration. (2025). Q&A on FDA regulation of intentional genomic alterations in animals. Center for Veterinary Medicine.
U.S. National Institutes of Health. (2024-2026). Gene editing research updates. National Human Genome Research Institute.
Navigation
© 2026 FyreIgnis Market Research. All rights reserved.
Legal
info@fyreignismarketresearch.com
India
