
Breaking the Clinical Bottleneck


The pharmaceutical industry stands at an inflection point. In 2024, the FDA issued more than 60 oncology approvals, including 11 first-in-class therapeutics, demonstrating unprecedented momentum in cancer drug development. Yet behind these achievements lies a critical transformation: the strategic integration of real-world evidence (RWE) into regulatory pathways. This shift is not merely procedural—it represents a fundamental reimagining of how we validate therapeutic benefit, compress development timelines, and ultimately accelerate patient access to life-saving treatments.
For pharmaceutical strategists navigating increasingly complex development landscapes, understanding and leveraging RWE has evolved from competitive advantage to strategic imperative.
The Clinical Trial Bottleneck: Quantifying the Challenge
Traditional randomized controlled trials (RCTs) remain the gold standard for establishing efficacy, but their limitations have become increasingly apparent in oncology development:
The Time-Cost-Patient Paradox
Clinical trials face three interconnected constraints:
Timeline Constraints: The average oncology drug development cycle spans 10-15 years from discovery to approval, with Phase III trials alone consuming 3-5 years.
Financial Burden: Development costs for oncology therapeutics have escalated dramatically, with industry estimates suggesting total costs exceeding $2.6 billion per approved drug when factoring in failure rates.
Patient Recruitment Challenges: Rare oncology indications face particularly acute challenges. For cancers affecting fewer than 200,000 patients annually, recruiting adequate trial populations becomes a rate-limiting factor in development timelines.
These constraints create a clinical bottleneck that delays patient access and increases development risk—precisely the challenges RWE is positioned to address.
The Real-World Evidence Revolution: From Theory to Practice
What Constitutes Real-World Evidence in Oncology?
Real-world evidence derives from analysis of real-world data (RWD) collected outside traditional clinical trial settings. In oncology, this encompasses:
Electronic Health Records (EHR): Patient demographics, treatment patterns, clinical outcomes
Claims and Billing Data: Treatment utilization, healthcare resource consumption
Patient Registries: Disease-specific longitudinal data
Patient-Generated Data: Quality of life measures, patient-reported outcomes
The FDA's Oncology Real-World Evidence Program, established in 2020, has completed over 200 reviews spanning drugs, biologics, and devices, demonstrating the maturation of RWE as a regulatory tool.
Statistical Snapshot: RWE Adoption in Oncology Approvals
Recent data reveals the accelerating integration of RWE into regulatory submissions:
Table 1: Real-World Evidence in FDA Labeling Expansions (2022-2024)
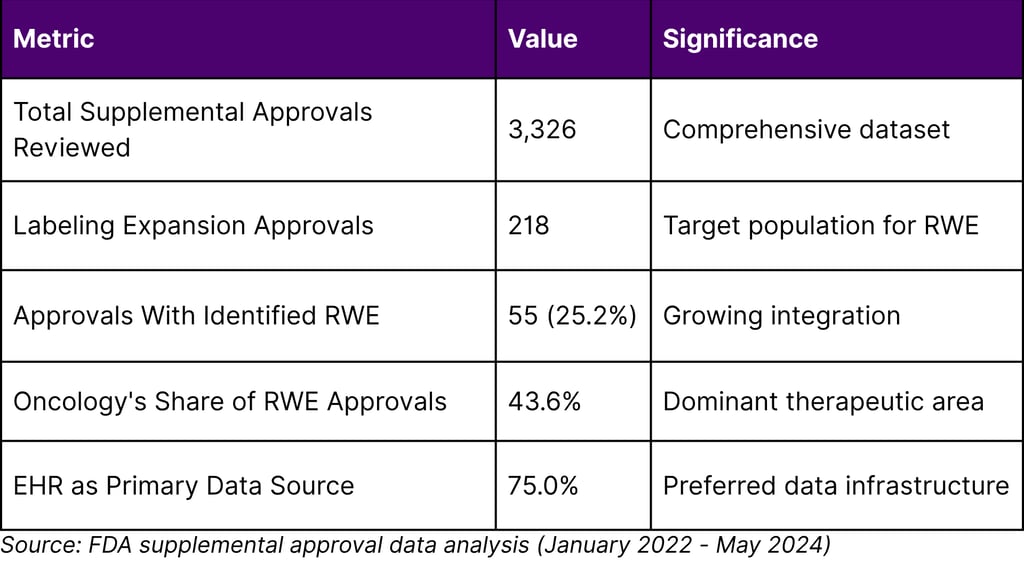
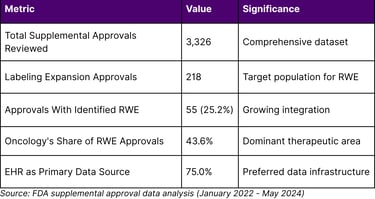
Among 218 labeling expansion approvals, 25.2% utilized real-world evidence, with oncology representing 43.6% of these RWE-supported approvals. The predominance of electronic health records as data sources (75%) underscores the importance of robust data infrastructure investments.
Figure 1: Real-World Evidence Study Design Distribution
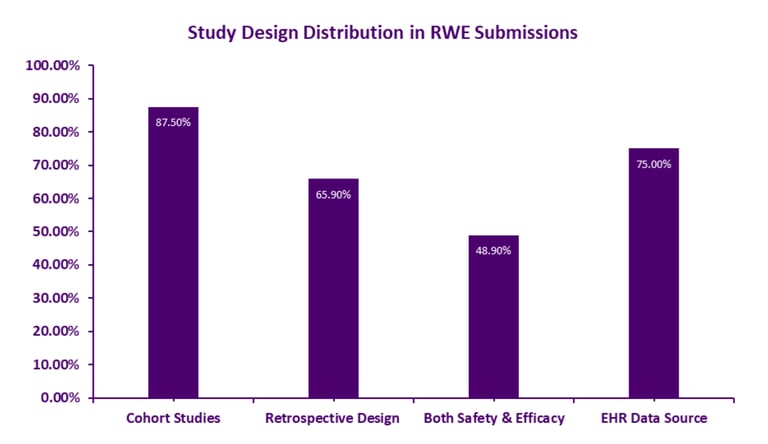
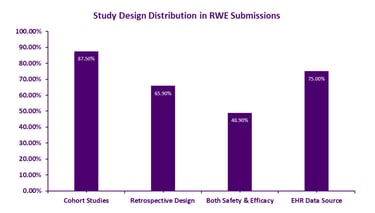
Strategic Use Cases: Where RWE Drives Regulatory Success
1. Project Renewal: Modernizing Legacy Therapeutics
Project Renewal, an FDA Oncology Center of Excellence initiative, reviews real-world data to update approved indications for older drugs. This program exemplifies RWE's value in lifecycle management.
Case Study: Durvalumab and Fludarabine
In December 2024, durvalumab received approval for limited-stage small cell lung cancer under Project Renewal, making it the first immunotherapy approved for this indication. Similarly, fludarabine phosphate received updated labeling for B-cell chronic lymphocytic leukemia, demonstrating how RWE can expand therapeutic utility of established agents.
Through 2024, Project Renewal facilitated approval of 15 NDA supplements for new indications or dosage regimens, with participation from 34 oncologists and 26 clinical fellows across 42 U.S. institutions.
2. Accelerated Approval Pathway: Balancing Speed and Rigor
The accelerated approval pathway leverages surrogate endpoints "reasonably likely to predict clinical benefit," with RWE playing an increasingly important role in confirmatory evidence.
Table 2: FDA Accelerated Approval Landscape (2024)
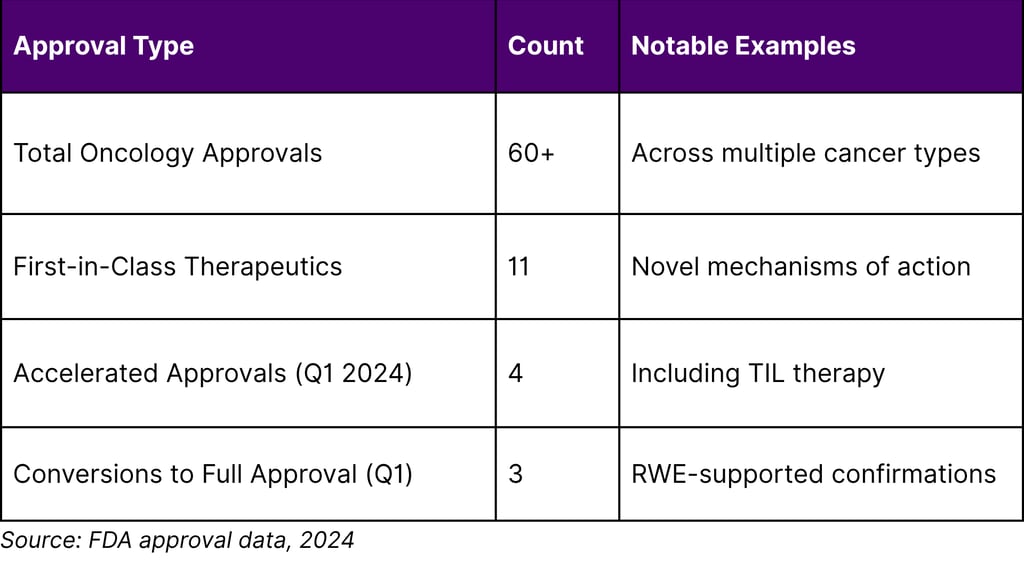
Critical Insight on Accelerated Approvals:
Analysis of cancer drugs granted accelerated approval between 2013-2017 revealed that 63% converted to regular approval, though only 43% demonstrated clinical benefit in confirmatory trials after five years. This finding underscores both the promise and the ongoing challenges in the accelerated approval pathway, highlighting the need for robust confirmatory evidence—where RWE can play a complementary role.
3. Rare Cancer Indications: Solving the Impossible Trial
For rare cancers where traditional RCTs may be infeasible, RWE offers a path forward.
Zenocutuzumab received accelerated approval for NRG1 fusion-positive non-small cell lung cancer and pancreatic adenocarcinoma, representing the first targeted therapy for tumors with NRG1 gene fusions. In populations this rare, RWE from expanded access programs and patient registries becomes essential for regulatory decision-making.
RWE has been particularly valuable for rare or orphan indications, supporting both original and supplementary oncology drug approvals through historical controls.
The FDA's Strategic Framework: Infrastructure Supporting RWE Integration
Key Initiatives Shaping the RWE Landscape
1. The QCARD Initiative
The Oncology Quality, Characterization and Assessment of Real-World Data (QCARD) initiative facilitates communication between sponsors and FDA reviewers to improve review efficiency for early RWD study proposals. This proactive framework helps sponsors design RWE studies that meet regulatory standards from inception.
2. The MoRE Glossary
In 2024, the FDA completed development of the Modernizing Research and Evidence (MoRE) Glossary through FDA-NIH collaboration, fostering consistent terminology in RWE submissions—a critical step for standardizing regulatory review.
3. Real-World Endpoints Development
The FDA collaborated with Friends of Cancer Research on evaluation of real-world tumor response derived from electronic health record data sources in patients with metastatic non-small cell lung cancer treated with chemotherapy, advancing the science of RWE endpoint validation.
FDA Oncology RWE Program Evolution (2020-2024)
Program Milestones:
2020 ║ Oncology RWE Program Established ║ Initial framework development
2021 ║ Project Renewal Launch ║ First legacy drug reviews
2022 ║ QCARD Initiative Implementation ║ Standardized data element requirements
2023 ║ MoRE Glossary Development ║ FDA-NIH terminology collaboration
2024 ║ 200+ Completed Reviews ║ Mature regulatory infrastructure ║ AI Initiative Launch for RWE analysis
Commercial Acceleration: The Strategic Value Proposition
Quantifying the RWE Advantage
For pharmaceutical strategists, RWE offers tangible benefits across the development lifecycle:
1. Timeline Compression
Reduced Enrollment Periods: Leveraging existing data eliminates months of patient recruitment
Parallel Evidence Generation: RWE can be collected concurrently with ongoing trials
Faster Regulatory Review: Well-designed RWE submissions aligned with QCARD principles receive more efficient review
2. Risk Mitigation
Early Signal Detection: RWE identifies safety signals and efficacy patterns before committing to pivotal trials
Population Representativeness: Real-world populations include the diversity often excluded from RCTs
Post-Approval Sustainability: RWE supports lifecycle management and label expansions
3. Cost Optimization
While specific cost data varies by program, strategic RWE deployment can:
Reduce per-patient costs in confirmatory trials
Enable smaller, targeted pivotal studies
Support multiple indication expansions from single data sources
Strategic Positioning Framework
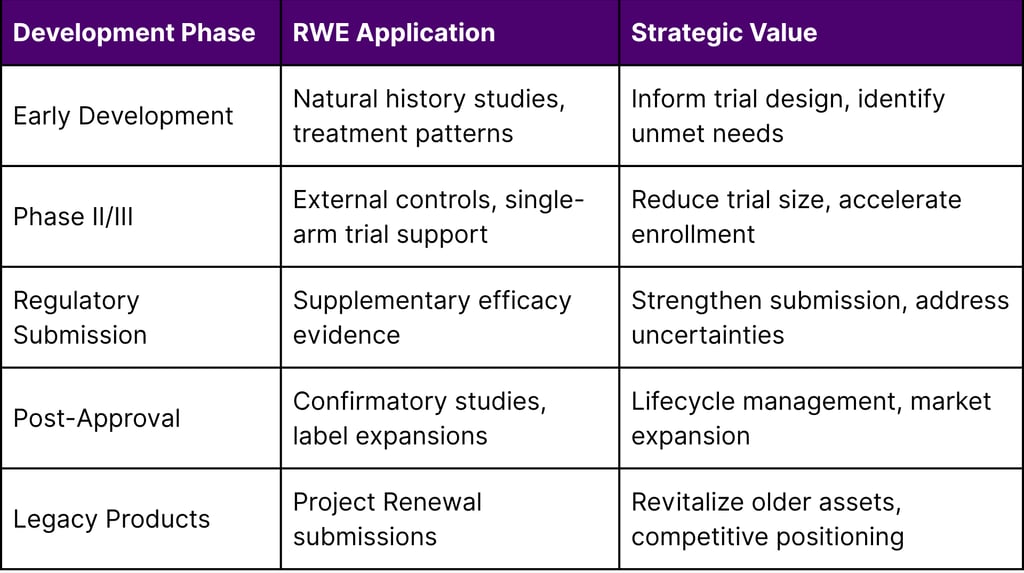
Table 3: RWE Strategic Application by Development Phase

Navigating the Challenges: Critical Success Factors
Methodological Rigor Requirements
FDA reviewers have cited small sample sizes, data quality issues, and methodological concerns in RWE submissions, highlighting the importance of rigorous study design.
Key Success Factors:
Data Quality Assurance
Comprehensive source verification
Missing data management protocols
Bias mitigation strategies
Appropriate Study Design
Clear research questions aligned with regulatory needs
Pre-specified analysis plans
Sensitivity analyses for robustness
Transparent Reporting
Detailed data source characterization
Explicit limitations discussion
Reproducible analytical methods
Regulatory Engagement Strategy
The Advancing RWE Program conducts meetings with sponsors during fiscal years 2023-2027 to identify approaches for generating RWE that meet regulatory requirements. Early regulatory engagement dramatically improves RWE study success rates.
Recommended Engagement Timeline:
Pre-IND Stage: Discuss RWE strategy and data sources
Phase II: Present preliminary RWE findings and confirmatory plans
NDA/BLA Preparation: Review RWE study protocols and preliminary results
Post-Approval: Align on RWE requirements for confirmatory studies
Future Trajectories: The Next Frontier in RWE Innovation
Emerging Technologies Reshaping RWE
1. Artificial Intelligence Integration
In 2024, the FDA OCE launched an oncology-specific AI initiative to advance understanding of AI applications in RWE generation and analysis. Machine learning algorithms promise to:
Extract structured data from unstructured EHR notes
Identify previously undetectable treatment response patterns
Enable predictive modeling for trial enrichment
2. Wearable Technology and Patient-Generated Data
The In4M project, a Yale-Mayo CERSI collaboration, aims to characterize longitudinal measurement characteristics of physical function assessments, including patient-reported outcomes, performance tests, and wearable data. This convergence of technology and patient experience data represents the next evolution in RWE sophistication.
3. Pragmatic Trial Integration
Project Pragmatica expanded in 2024 to include broader evidence generation efforts across HHS and public-private partnerships, including the Project 5 in 5 crowdsourcing initiative for pragmatic trial concepts—blending traditional trial rigor with real-world practicality.
Global Harmonization Efforts
OCE conducted collaboration visits in late 2024 with Health Canada, European regulators including Belgium's FAMHP, UK's MHRA, and Switzerland's Swissmedic, focusing on regulatory cooperation in oncology. These international partnerships suggest growing global alignment on RWE standards, creating opportunities for multi-regional RWE strategies.
Strategic Imperatives for Pharmaceutical Leaders
Building RWE Capabilities: A Roadmap
Phase 1: Foundation (Months 1-6)
Audit existing data infrastructure and gaps
Establish cross-functional RWE governance
Develop relationships with data partners and academic centers
Initiate FDA engagement through Advancing RWE Program
Phase 2: Pilot Implementation (Months 7-18)
Execute proof-of-concept RWE study
Validate data quality and analytical methodologies
Present preliminary findings to regulatory agencies
Refine internal processes based on learnings
Phase 3: Strategic Integration (Months 19+)
Embed RWE in standard development planning
Build therapeutic area-specific RWE playbooks
Expand data partnerships and infrastructure
Leverage RWE for competitive intelligence and market access
Key Performance Indicators
Monitor these metrics to assess RWE program maturity:
Regulatory Success Rate: Percentage of RWE submissions accepted by FDA
Timeline Impact: Months saved in development timelines
Label Expansion Velocity: Rate of new indication approvals leveraging RWE
Data Quality Metrics: Completeness, accuracy, and timeliness of RWD sources
Stakeholder Engagement: Number of regulatory interactions and collaborations
Conclusion: The Competitive Imperative
The integration of real-world evidence into oncology drug development has transitioned from experimental to essential. With 32 notable precision oncology therapeutic approvals in 2024, including the first tumor-agnostic antibody-drug conjugate approval, the FDA has demonstrated its commitment to evidence modernization.
For pharmaceutical strategists, the question is no longer whether to invest in RWE capabilities, but how quickly and strategically to build them. Organizations that master the generation, analysis, and regulatory submission of high-quality real-world evidence will capture disproportionate value through:
Accelerated development timelines
Reduced late-stage development risk
Enhanced lifecycle management capabilities
Stronger competitive positioning in crowded therapeutic spaces
The clinical bottleneck is breaking. The strategic winners will be those who recognized RWE not as a regulatory checkbox, but as a transformative tool for drug development acceleration and commercial success.
Frequently Asked Questions (FAQs)
Q1: What is the primary difference between real-world data (RWD) and real-world evidence (RWE)?
A: Real-world data refers to the raw information collected from sources outside traditional clinical trials (EHRs, claims, registries). Real-world evidence is the clinical evidence derived from analysis and interpretation of that data. Think of RWD as the raw material and RWE as the refined product used for regulatory decision-making.
Q2: Can real-world evidence completely replace randomized controlled trials in oncology?
A: No. RWE complements rather than replaces RCTs. The FDA views RWE as most appropriate for supplementary evidence, rare disease indications where RCTs are infeasible, and post-approval confirmatory studies. For most novel therapeutic approvals, RCTs remain the primary evidence standard, with RWE providing supporting data.
Q3: What percentage of oncology drug approvals currently utilize real-world evidence?
A: Based on recent data, approximately 25% of labeling expansion approvals incorporate real-world evidence, with oncology representing 44% of all RWE-supported approvals. However, adoption rates are increasing rapidly as regulatory frameworks mature and data infrastructure improves.
Q4: How long does it typically take to generate regulatory-grade real-world evidence?
A: Timeline varies significantly based on study complexity, data availability, and regulatory endpoints. Simple retrospective cohort studies leveraging existing EHR data may take 6-12 months, while prospective registry studies requiring new data collection can extend 2-3 years or longer. Early FDA engagement through the Advancing RWE Program can help optimize timelines.
Q5: What are the most common reasons FDA rejects real-world evidence submissions?
A: Key rejection factors include inadequate data quality, small sample sizes, methodological flaws (selection bias, confounding), unclear research questions, and insufficient documentation of data sources. Following QCARD guidance and securing FDA feedback on protocols before study initiation dramatically improves success rates.
Q6: Which data sources are most valuable for oncology real-world evidence?
A: Electronic health records represent 75% of RWE data sources and are particularly valuable for oncology due to their comprehensive clinical information. Claims data provides treatment patterns and resource utilization. Disease-specific registries offer depth in particular therapeutic areas. The optimal approach often combines multiple complementary data sources.
Q7: How does Project Renewal differ from standard supplemental approval pathways?
A: Project Renewal specifically targets older oncology drugs (often decades-old) where substantial real-world experience has accumulated but labels haven't been updated. The program leverages external oncologists to review published literature and real-world data, providing a streamlined pathway to modernize labeling based on contemporary evidence and practice patterns.
Q8: What role will artificial intelligence play in future real-world evidence generation?
A: AI will likely transform RWE through automated data extraction from unstructured clinical notes, pattern recognition in large datasets, predictive modeling for patient outcomes, and quality assurance processes. The FDA's 2024 launch of an oncology-specific AI initiative signals regulatory openness to these innovations, though validation and transparency requirements will be critical.
Q9: How should smaller biotechnology companies approach RWE strategy given limited resources?
A: Focus resources on early regulatory engagement (Advancing RWE Program, QCARD framework), strategic data partnerships rather than building infrastructure, and targeted RWE studies addressing specific regulatory questions. Consider academic collaborations and the FDA's Project Catalyst initiative designed specifically for small companies and academic developers.
Q10: What impact does accelerated approval pathway performance have on RWE strategy?
A: The finding that only 43% of accelerated approvals demonstrated clinical benefit in confirmatory trials highlights the importance of robust post-approval evidence generation. This creates both opportunity (RWE can support confirmatory evidence) and imperative (higher scrutiny of post-approval commitments). Companies should plan RWE strategies that specifically address confirmatory study requirements at the time of accelerated approval.
References
FDA. (2024). "Ongoing Clinical Oncology Projects 2024 - Oncology Center of Excellence 2024 Annual Report." U.S. Food and Drug Administration.
American Association for Cancer Research. (2025). "FDA Approvals in Oncology October-December 2024." Cancer Research Catalyst.
FDA. (2024). "OCE Regulatory Programs 2024 - Oncology Center of Excellence 2024 Annual Report." U.S. Food and Drug Administration.
Liu, I.T.T., Cliff, E.R.S., et al. (2024). "Real-World Evidence in FDA Approvals for Labeling Expansion of Small Molecules and Biologics." Therapeutic Innovation & Regulatory Science, 59, 982-992.
Mishra-Kalyani, P.S., et al. (2020). "Use of Real-World Evidence to Support FDA Approval of Oncology Drugs." Journal of Clinical Oncology. PubMed, 23(10), 1358–1365.
FDA. "Oncology Real World Evidence Program." U.S. Food and Drug Administration.
FDA. "Oncology Quality, Characterization and Assessment of Real-World Data (QCARD) Initiative." U.S. Food and Drug Administration.
FDA. "Advancing Real-World Evidence Program." U.S. Food and Drug Administration.
American Association for Cancer Research. (2024). "Fewer Than Half of Accelerated Approval Drugs Showed Clinical Benefit in Confirmatory Trials After Five Years." AACR News Release.
Agrawal, S., Park, E., & Kluetz, P.G. (2025). "FDA approvals in 2024: new options for patients across cancer types and therapeutic classes." Nature Reviews Clinical Oncology, 22, 457-458.
American Association for Cancer Research. (2024). "FDA Approvals in Oncology: January-March 2024." Cancer Research Catalyst.
American Association for Cancer Research. (2025). "FDA Approvals in Oncology: April-June 2025." Cancer Research Catalyst.
FDA. "Project Renewal - FDA Oncology Center of Excellence." U.S. Food and Drug Administration.
FDA. (2024). "Oncology Research and Development 2024 - Oncology Center of Excellence 2024 Annual Report." U.S. Food and Drug Administration.
Navigation
© 2026 FyreIgnis Market Research. All rights reserved.
Legal
info@fyreignismarketresearch.com
India
