
Gene Editing Enters The Clinic


The gene editing landscape is undergoing a transformative shift as CRISPR-based therapies transition from experimental platforms to approved clinical treatments. Following the FDA's landmark December 2023 approval of Casgevy, the first CRISPR/Cas9-based therapy for sickle cell disease and transfusion-dependent beta-thalassemia, the field has accelerated toward multiple regulatory milestones anticipated in 2026. With approximately 250 clinical trials involving gene-editing therapeutic candidates currently monitored globally and more than 150 trials actively recruiting participants, the competitive intelligence landscape demands close attention to emerging base editing platforms, next-generation delivery systems, and novel regulatory pathways.
This comprehensive analysis examines the current state of CRISPR 3.0 technologies, base editing innovations, pivotal clinical trial data, and the FDA's newly introduced plausible mechanism pathway that promises to revolutionize personalized gene therapy approvals. Investment opportunities and competitive positioning insights are provided for stakeholders navigating this rapidly evolving therapeutic domain.
INTRODUCTION: THE CLINICAL TRANSLATION OF GENE EDITING
Gene editing technology has progressed from theoretical concept to clinical reality with unprecedented speed. The FDA's approval of Casgevy in December 2023 marked a watershed moment not only as the first CRISPR/Cas9-based therapy to receive regulatory clearance, but as validation of an entirely new therapeutic modality capable of addressing the root genetic causes of previously intractable diseases.
Casgevy, developed jointly by Vertex Pharmaceuticals and CRISPR Therapeutics, targets sickle cell disease and transfusion-dependent beta-thalassemia by editing patients' hematopoietic stem cells ex vivo to increase fetal hemoglobin production. Clinical trial data demonstrated remarkable efficacy: 93.5% of evaluable sickle cell disease patients achieved freedom from severe vaso-occlusive crises for at least 12 consecutive months. As of December 2025, Casgevy has received approval in multiple jurisdictions including the United States, United Kingdom, European Union, Switzerland, Canada, Bahrain, Saudi Arabia, and the United Arab Emirates.
THE CURRENT CRISPR CLINICAL TRIALS LANDSCAPE
Global Trial Statistics
As of December 2025, the gene editing clinical trial ecosystem has expanded substantially. According to data from ClinicalTrials.gov and comprehensive monitoring platforms, approximately 250 clinical trials involving gene-editing therapeutic candidates are currently registered globally, with more than 150 trials actively recruiting participants. This represents significant growth from the 84 CRISPR-based clinical trials identified in comprehensive reviews published through 2023.
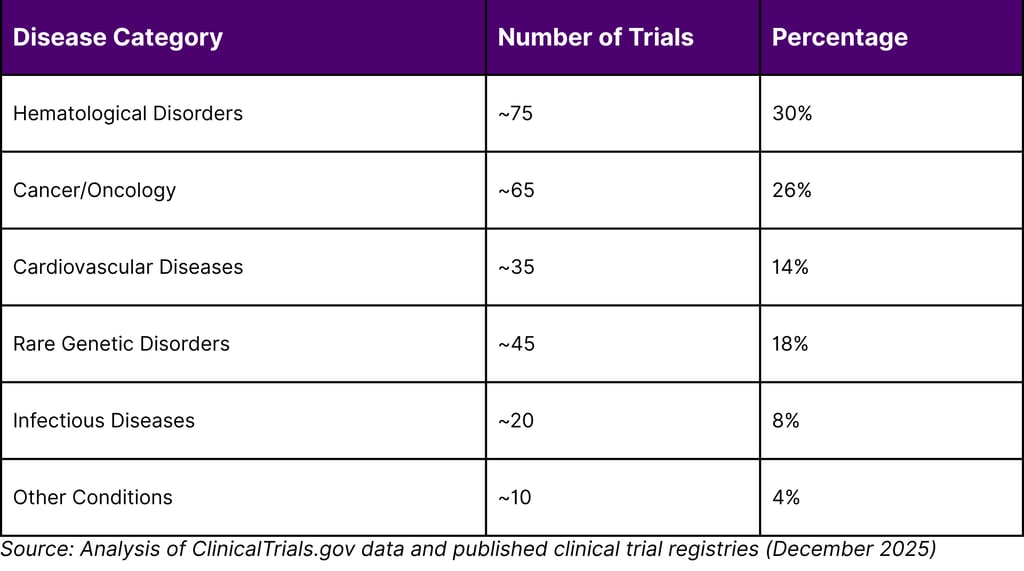
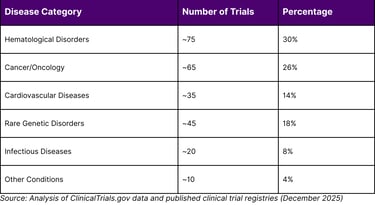
The therapeutic areas encompass a broad spectrum of conditions: hematological disorders continue to dominate with the majority of Phase 3 trials, followed by oncology applications (particularly CAR-T cell therapies), cardiovascular diseases, rare genetic disorders, and infectious diseases. Notably, 35% of trials target genetic diseases while 65% address non-genetic conditions, reflecting CRISPR's versatility beyond inherited disorders.
TABLE 1: CRISPR CLINICAL TRIALS BY DISEASE CATEGORY (2025 DATA)
BASE EDITING: THE NEXT GENERATION OF PRECISION
Base editing represents a significant advancement over traditional CRISPR-Cas9 technology. Unlike conventional gene editing that creates double-stranded DNA breaks, base editors make precise single-nucleotide changes without severing both DNA strands. This approach reduces certain safety risks associated with chromosomal rearrangements and unwanted insertions or deletions.
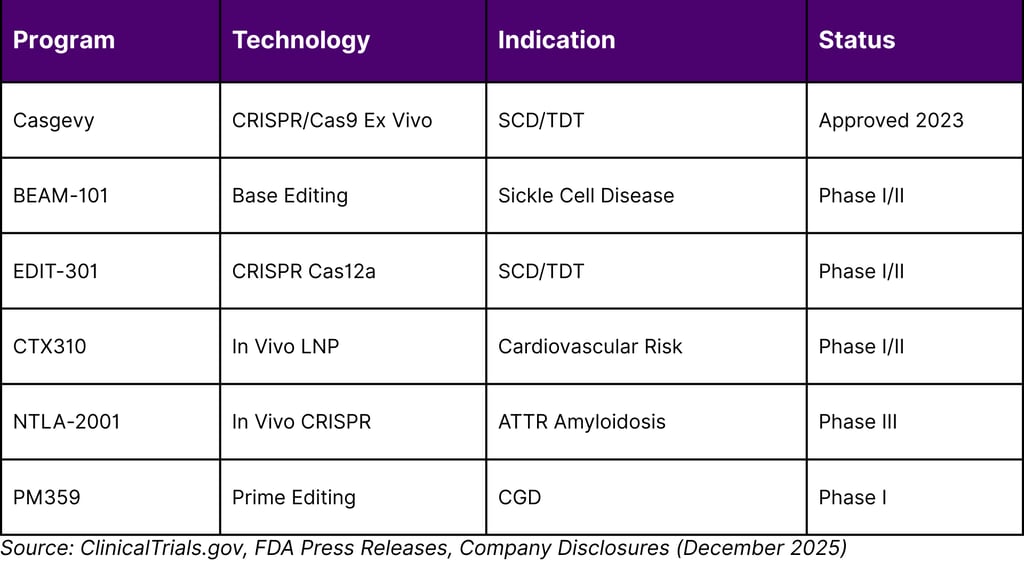
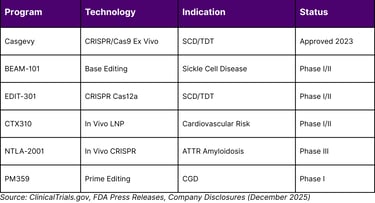
Beam Therapeutics has emerged as a leader in base editing with its BEAM-101 program for sickle cell disease. In their Phase I/II BEACON trial, Beam dosed at least 17 adult patients as of mid-2025, with data from the first cohorts showing robust results. In the highest dose cohort, approximately 90% of alpha-1 antitrypsin protein in participants' blood samples was the healthy version by day 14, and this result was sustained at day 28. The FDA has cleared Beam to begin treating adolescent participants, positioning BEAM-101 for potential regulatory milestones in 2026.
Base editing's advantages include mimicking natural genetic variations associated with hereditary persistence of fetal hemoglobin, potentially offering improved safety profiles compared to approaches that create double-strand breaks. Clinical data presented at the American Society of Hematology meeting in December 2024 showed greater than 60% HbF induction and less than 40% Hemoglobin S reduction in all seven evaluated patients, with resolution of anemia across the cohort.
Prime Editing: Direct Mutation Correction
Prime editing, another advanced gene editing modality, enables precise insertions, deletions, and all 12 possible base-to-base conversions without requiring double-stranded breaks or donor DNA templates. Prime Medicine announced positive results in May 2025 from their trial in chronic granulomatous disease, marking the first clinical data demonstrating prime editing's efficacy and safety in humans. The trial participant showed no serious adverse events and robust restoration of protein function needed for normal white blood cell activity.
IN VIVO GENE EDITING: ELIMINATING EX VIVO CHALLENGES
A critical limitation of current approved CRISPR therapies like Casgevy is the requirement for ex vivo editing. Patients must undergo myeloablative conditioning with high-dose chemotherapy to prepare for reinfusion of edited cells a demanding process that limits patient eligibility and poses significant risks. In vivo gene editing approaches promise to overcome these barriers by delivering editing components directly to target tissues.
Lipid nanoparticle (LNP) delivery systems have emerged as the leading platform for in vivo CRISPR delivery to the liver. Multiple companies including CRISPR Therapeutics, Beam Therapeutics, and Verve Therapeutics are advancing LNP-delivered base editing programs targeting cardiovascular risk factors. In November 2025, Cleveland Clinic presented Phase 1 results for CTX310, a CRISPR-Cas9 therapy targeting the ANGPTL3 gene. One-time infusion resulted in approximately 50% reduction in LDL cholesterol and 55% reduction in triglycerides within two weeks, with sustained effects through at least 60 days of follow-up and no serious treatment-related adverse events.
NTLA-2001, developed by Intellia Therapeutics for transthyretin amyloidosis, has advanced to Phase 3 trials after demonstrating greater than 90% reduction in pathogenic TTR protein levels with clinical metrics indicating disease stabilization. These successes validate the in vivo approach and position multiple programs for regulatory readouts in 2026-2027.
TABLE 2: LEADING GENE EDITING PROGRAMS APPROACHING REGULATORY MILESTONES
REGULATORY INNOVATION: THE PLAUSIBLE MECHANISM PATHWAY
In November 2025, FDA officials Dr. Martin Makary and Dr. Vinay Prasad unveiled a transformative regulatory framework: the plausible mechanism pathway for individualized therapies. This approach shifts regulatory focus from approving each individual customized therapy to clearing therapeutic platforms based on established biological mechanisms. Rather than requiring extensive efficacy studies for each patient-specific treatment, the FDA will grant approvals where pharmacologic effect aligns with biologic plausibility and observed clinical outcomes.
The pathway's development was catalyzed by the case of Kyle Patrick Muldoon, Jr. (KJ), an infant diagnosed with severe CPS1 deficiency. Researchers at Children's Hospital of Philadelphia and the University of Pennsylvania developed a customized CRISPR therapy within seven months. The FDA processed the single-patient expanded-access application in one week, enabling rapid manufacturing and infusion. The treatment has successfully stabilized KJ's condition, allowing him to tolerate high dietary protein while reducing his nitrogen-scavenger drug dosage by half, with no severe adverse effects reported through May 2025.
This regulatory innovation is expected to dramatically accelerate the development timeline for rare disease therapies. Initial approvals under the platform technology designation framework could arrive within three years for genetic diseases treatable by editing liver or blood stem cells. Academic institutions including University of Pennsylvania-CHOP, University of Wisconsin, and IGI-UCSF are planning umbrella trials for 2026 that would enroll children with specific clinical syndromes regardless of their exact mutation, provided they share a common biological mechanism.
CRISPR-EDITED CAR-T: TRANSFORMING CANCER TREATMENT
CRISPR technology is revolutionizing chimeric antigen receptor T-cell (CAR-T) therapy by enabling development of allogeneic (off-the-shelf) products. Traditional autologous CAR-T therapies, while effective, require time-consuming and expensive manufacturing processes for each individual patient. CRISPR-edited allogeneic CAR-T cells can be manufactured at scale from healthy donors, potentially reducing costs and improving accessibility.
CRISPR Therapeutics has advanced multiple allogeneic CAR-T candidates into clinical trials. CTX110, targeting CD19-positive malignancies, and CTX130, targeting CD70-positive cancers, have shown favorable results in B-cell and T-cell lymphomas and renal cell carcinoma. In one Phase 1 trial, CRISPR-modified CAR-T cells targeting CD19 with attenuated PD-1 expression showed complete remission in 87.5% of patients and partial remission in 12.5%, with no serious adverse events during 12 months of follow-up.
However, challenges remain. Some allogeneic CAR-T products have encountered T cell exhaustion leading to loss of response and reduced efficacy, particularly in high tumor burden settings. Ongoing research focuses on additional genetic modifications to enhance persistence and functionality of edited cells.
MARKET DYNAMICS AND INVESTMENT LANDSCAPE
The gene editing sector faces a complex market environment in 2026. While technological breakthroughs continue to accelerate, financial pressures have intensified. Reduced venture capital investment combined with the high cost of clinical trials has led to strategic reorganizations across the industry. Multiple CRISPR-focused companies have announced workforce reductions and portfolio narrowing to focus resources on advancing lead programs toward commercialization rather than building broad pipelines.
Casgevy's commercial launch provides important market validation data. CRISPR Therapeutics reported through Q3 2025 that 25 authorized treatment centers had initiated more than 5 patients each, with at least one center in each region initiating 20 or more patients. Vertex Pharmaceuticals projects Casgevy revenue exceeding $100 million in 2025 with significant growth expected in 2026. At $2.2 million for the one-time treatment, Casgevy represents the highest-priced therapy in the current market, though its curative potential may justify the cost compared to lifetime management of chronic conditions.
Reimbursement agreements are expanding globally. Italy, which has approximately 5,000 people aged 12 and older with transfusion-dependent beta-thalassemia and approximately 2,300 with sickle cell disease, reached a reimbursement agreement in September 2025. Such agreements are critical for market access and will influence the commercial viability of future gene editing therapies.
SAFETY CONSIDERATIONS AND REGULATORY VIGILANCE
Safety remains paramount as gene editing transitions from controlled trials to broader clinical use. The tragic deaths of two teenage Duchenne muscular dystrophy patients who received Sarepta's Elevidys gene therapy in 2025 highlighted ongoing risks associated with high-dose viral vector delivery. While these events were attributed to acute liver failure potentially triggered by the AAV vector rather than the therapeutic transgene, they underscore the importance of careful patient selection, dosing optimization, and long-term safety monitoring.
For CRISPR therapies specifically, key safety considerations include off-target editing effects, chromosomal rearrangements from double-strand breaks, immune responses to Cas proteins, and potential long-term genotoxicity. Base editing and prime editing platforms that avoid double-strand breaks may offer improved safety profiles, but long-term follow-up data remain limited. All approved CRISPR therapies require patients to be followed in long-term studies to evaluate safety and sustained effectiveness.
Lyfgenia, the second FDA-approved gene therapy for sickle cell disease, carries a black box warning for hematologic malignancy risk. This highlights that even approved therapies require ongoing surveillance and that benefit-risk assessments must account for potential serious long-term complications.
2026 OUTLOOK: ANTICIPATED REGULATORY MILESTONES
Multiple gene editing programs are positioned to reach critical regulatory decision points in 2026. Based on current clinical trial timelines and company guidance, the following milestones are anticipated:
Early 2026: Phase I/II data updates from base editing programs (BEAM-101, VERVE-201) targeting cardiovascular risk factors are expected. Positive results could support advancement to pivotal trials and potential breakthrough therapy designations from the FDA.
Mid-2026: CRISPR Therapeutics plans to initiate clinical trials for their alpha-1 antitrypsin deficiency program. Prime Medicine anticipates beginning an AATD trial. Umbrella trials under the plausible mechanism pathway for urea cycle disorders and severe T cell dysfunction are planned to begin enrollment.
Late 2026: Potential regulatory submissions for programs that have completed Phase II/III trials. NTLA-2001 for ATTR amyloidosis, currently in Phase 3, could approach regulatory filing depending on data maturity. Additional indications for approved platforms may receive expanded approval.
The FDA's implementation of the plausible mechanism pathway will be closely watched. Initial approvals under this framework would establish important precedents for personalized medicine and could dramatically expand the addressable rare disease population.
COMPETITIVE INTELLIGENCE: STRATEGIC POSITIONING
The competitive landscape for gene editing is characterized by strategic differentiation across multiple dimensions: editing modality (CRISPR-Cas9, Cas12, base editing, prime editing), delivery approach (ex vivo vs. in vivo), target tissues (blood, liver, muscle, eye, central nervous system), and therapeutic areas.
Companies pursuing ex vivo editing of hematopoietic stem cells face the challenge of competing against Casgevy's established efficacy and safety profile. Differentiation strategies include improved conditioning regimens, enhanced cell engraftment, or targeting patient populations unable to tolerate current protocols. Base editing approaches offer theoretical safety advantages that may prove compelling in head-to-head comparisons.
In vivo delivery platforms represent a blue ocean opportunity. Success in cardiovascular indications could establish proof-of-concept for LNP-delivered gene editing at scale, potentially applicable to numerous additional targets. However, competition is intensifying as multiple companies advance similar approaches. Intellectual property around editing components, delivery formulations, and specific target sequences will be crucial.
For rare disease programs, the plausible mechanism pathway creates both opportunities and competitive pressures. Academic-led initiatives may compete directly with commercial entities, particularly if non-profit models can demonstrate comparable efficacy at lower cost. Strategic partnerships between pharmaceutical companies and academic centers may emerge as an optimal structure.
CONCLUSION
Gene editing has definitively entered the clinical mainstream. The field's rapid evolution from Casgevy's approval in December 2023 to the current landscape of 250+ clinical trials demonstrates extraordinary momentum. Base editing and prime editing technologies are maturing beyond proof-of-concept toward potential commercial products. In vivo delivery systems are achieving clinical validation. Most significantly, the FDA's plausible mechanism pathway promises to transform rare disease treatment by enabling platform-based approvals rather than requiring individual therapy validation.
As 2026 unfolds, multiple regulatory readouts will determine which programs transition from promising candidates to approved therapies. Stakeholders must monitor clinical trial results, regulatory guidance documents, reimbursement decisions, and competitive positioning to navigate this rapidly evolving landscape. The next 12-18 months will be critical in determining whether the promise of CRISPR 3.0 translates into broad therapeutic impact.
FREQUENTLY ASKED QUESTIONS
Q1: What is the difference between CRISPR-Cas9 and base editing?
CRISPR-Cas9 creates double-stranded breaks in DNA to disable or modify genes, while base editing makes precise single-nucleotide changes without severing both DNA strands. Base editing may offer improved safety by reducing risks of chromosomal rearrangements and unintended insertions or deletions.
Q2: How many CRISPR therapies are currently approved by the FDA?
As of January 2026, the FDA has approved two gene therapies for sickle cell disease: Casgevy (CRISPR-based) and Lyfgenia (lentiviral vector-based). Casgevy is the only FDA-approved therapy utilizing CRISPR/Cas9 technology. It is approved for both sickle cell disease and transfusion-dependent beta-thalassemia.
Q3: What is the FDA's plausible mechanism pathway?
The plausible mechanism pathway is a new regulatory framework announced in November 2025 that enables approval of individualized therapies based on established biological mechanisms rather than requiring extensive efficacy studies for each patient-specific treatment. It focuses on therapeutic platforms and allows faster approvals when pharmacologic effects align with biological plausibility and observed clinical outcomes.
Q4: What are the main safety concerns with CRISPR therapies?
Key safety considerations include off-target editing effects, chromosomal rearrangements from double-strand breaks, immune responses to Cas proteins, potential long-term genotoxicity, and risks from viral vector delivery systems. Lyfgenia carries a black box warning for hematologic malignancy risk. All approved CRISPR therapies require long-term safety monitoring.
Q5: How much does Casgevy cost?
Casgevy is priced at $2.2 million for the one-time treatment, making it one of the highest-priced therapies currently available. However, as a potentially curative therapy, cost-effectiveness analyses compare this one-time cost against the lifetime costs of managing chronic sickle cell disease or transfusion-dependent beta-thalassemia.
Q6: What is the difference between ex vivo and in vivo gene editing?
Ex vivo editing involves removing cells from a patient, editing them in the laboratory, and reinfusing them after myeloablative conditioning. In vivo editing delivers gene editing components directly to target tissues within the body, potentially eliminating the need for chemotherapy conditioning and enabling treatment of tissues that cannot be easily removed and replaced.
Q7: How many clinical trials are currently investigating CRISPR therapies?
As of December 2025, approximately 250 clinical trials involving gene-editing therapeutic candidates are registered globally, with more than 150 trials actively recruiting participants. The trials span multiple therapeutic areas including hematological disorders, oncology, cardiovascular diseases, rare genetic disorders, and infectious diseases.
Q8: What are umbrella trials in the context of gene editing?
Umbrella trials enroll patients with a specific clinical syndrome regardless of their exact mutation, provided they share a common biological mechanism. Under the FDA's plausible mechanism pathway, these trials enable testing of platform technologies across multiple genetic variants, accelerating development for rare diseases where individual mutations affect very small patient populations.
REFERENCES
Cetin, B., Erendor, F., Eksi, Y. E., Sanlioglu, A. D., & Sanlioglu, S. (2025). Advancing CRISPR genome editing into gene therapy clinical trials: progress and future prospects. Expert Reviews in Molecular Medicine, 27, e16.
Cleveland Clinic. (2025, November 8). Cleveland Clinic first-in-human trial of CRISPR gene-editing therapy shown to safely lower cholesterol and triglycerides. Cleveland Clinic Newsroom.
CRISPR Medicine News. (2025). Overview CRISPR clinical trials 2026.
CRISPR Therapeutics. (2025, November 10). CRISPR Therapeutics provides business update and reports third quarter 2025 financial results [Press release].
Health Advances. (2025). 2025 marks technology, regulatory, and disease area shifts in cell and gene therapy.
Innovative Genomics Institute. (2025, July 9). CRISPR clinical trials: A 2025 update.
Musunuru, K., et al. (2025). In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature, 593, 258-262. Referenced in: Bharti, A., & Mudge, J. (2025). Therapeutic applications of CRISPR-Cas9 gene editing. Frontiers in Genome Editing, 7, 1724291.
U.S. Food and Drug Administration. (2023, December 8). FDA approves first gene therapies to treat patients with sickle cell disease [Press release].
Wang, D., Zhang, F., & Gao, G. (2023). Current trends of clinical trials involving CRISPR/Cas systems. Frontiers in Medicine, 10, 1292452.
Navigation
© 2026 FyreIgnis Market Research. All rights reserved.
Legal
info@fyreignismarketresearch.com
India
