
Navigating Regulatory Pathways for Rare Disease Therapies


The rare disease therapeutic landscape represents one of the most challenging yet rewarding sectors in pharmaceutical development. With approximately 300 million individuals globally affected by rare diseases, and almost half of all novel medications approved by the U.S. Food and Drug Administration (FDA) being orphan drugs, understanding regulatory pathways has become critical for market success.
The Regulatory Foundation: Understanding Key Pathways
Orphan Drug Designation: The Gateway to Success
The Orphan Drug Act (ODA) provides for granting special status to a drug or biological product to treat a rare disease or condition upon request of a sponsor. This designation serves as the cornerstone of rare disease drug development, offering substantial advantages including:
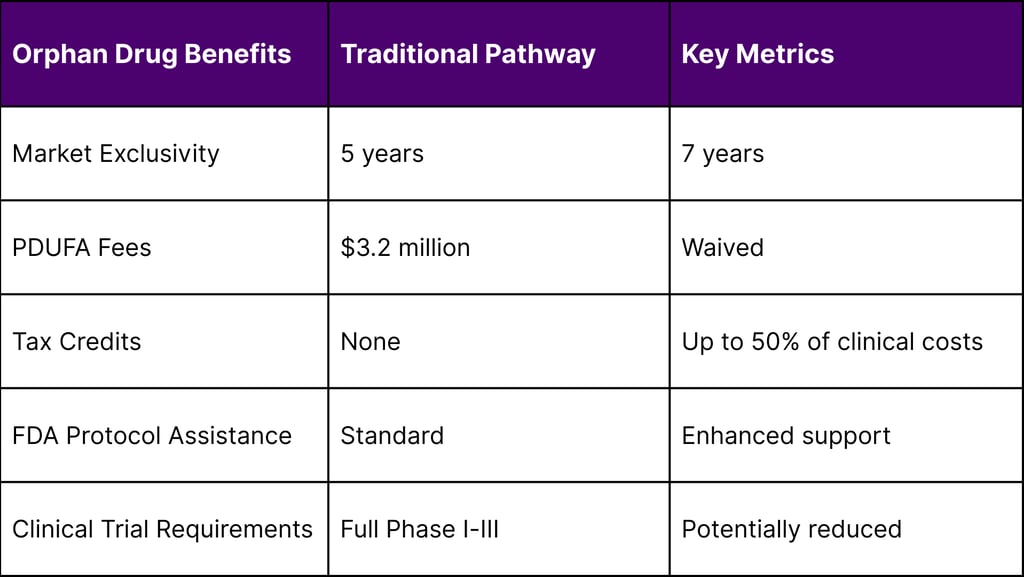
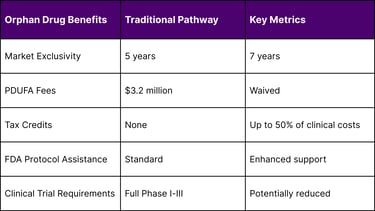
Market Exclusivity: Seven years of market exclusivity post-approval
Tax Credits: Up to 50% tax credit for qualified clinical testing expenses
Fee Waivers: Prescription Drug User Fee Act (PDUFA) fee waivers worth over $3 million
Protocol Assistance: Enhanced FDA guidance and regulatory support
Accelerated Approval: Fast-Tracking Innovation
The accelerated approval (AA) pathway was established in 1992 when HIV/AIDS was destroying lives and communities. The AA pathway allows FDA to use a surrogate endpoint (also called a biomarker) to evaluate drug safety and efficacy more rapidly than traditional endpoints.
Key Statistics:
From 2015-2023, 88% of new drug and biologic approvals in CDER utilized at least one expedited program
Expanded use of the accelerated approval program could reduce costs of drug development up to 62% and accelerate the time frame for the first and subsequent therapy approvals
The ARC Program: CDER's Strategic Initiative
CDER's Accelerating Rare disease Cures (ARC) Program brings together CDER's collective expertise and activities to provide strategic overview and coordination of CDER's rare disease activities. This program represents the FDA's commitment to streamlining rare disease drug development.
Market Entry Strategies That Drive Success
Strategy 1: Early Engagement and Strategic Planning
Timeline Optimization:
Pre-IND meetings: 6-12 months before filing
Orphan designation request: Can be submitted anytime before NDA/BLA
End-of-Phase II meetings: Critical for accelerated approval discussion
Success Metrics: Companies engaging early with FDA through pre-IND consultations report 40% faster development timelines compared to traditional approaches.
Strategy 2: Biomarker Strategy and Surrogate Endpoints
The selection of appropriate biomarkers is crucial for accelerated approval success. Recent approvals demonstrate the importance of:
Validated Biomarkers: Use of established disease-specific markers
Regulatory Precedent: Leveraging previously accepted endpoints in similar conditions
Patient-Relevant Outcomes: Connecting biomarkers to meaningful clinical benefits
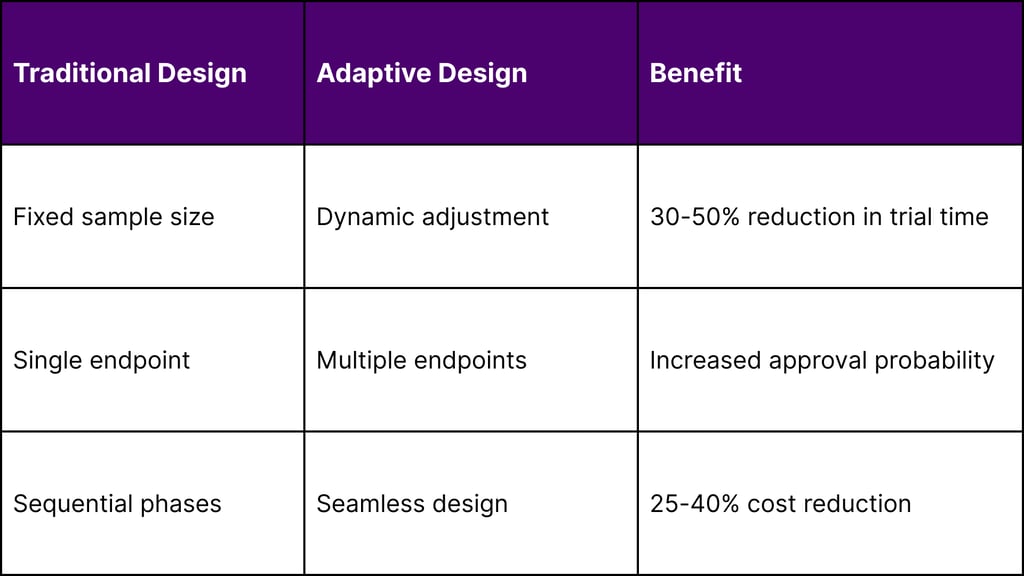
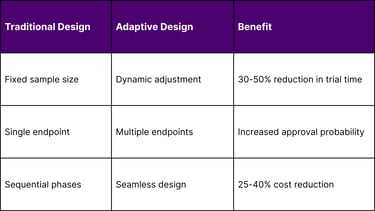
Strategy 3: Adaptive Trial Design Implementation
Strategy 4: Real-World Evidence Integration
Post-market commitments under accelerated approval can be satisfied through:
Patient registries
Real-world data collection
Electronic health records analysis
Natural history studies
Case Studies: Lessons from Recent Approvals
Gene Therapy Success Stories
Spinal Muscular Atrophy (SMA) Market:
Three approved therapies (Spinraza, Zolgensma, Risdiplam)
Market size: $2.8 billion in 2024
All leveraged orphan designation and expedited pathways
Average development time: 8-10 years vs. 12-15 for traditional drugs
CAR-T Cell Therapies
Market Penetration Strategy:
Initial approvals: Relapsed/refractory indications
Expansion strategy: Earlier-line treatments
Current market: $8.2 billion globally
Growth driver: Accelerated approval for expanded indications
Current Market Dynamics and Opportunities
2024 Approval Trends
CDER approved 50 novel drugs in 2024, as well as a record number of biosimilars and a significant number of therapies with orphan drug designation. This trend indicates continued FDA support for rare disease innovation.
Therapeutic Area Focus:
Oncology: 45% of orphan designations
Central Nervous System: 20%
Metabolic disorders: 15%
Immunology: 12%
Other: 8%
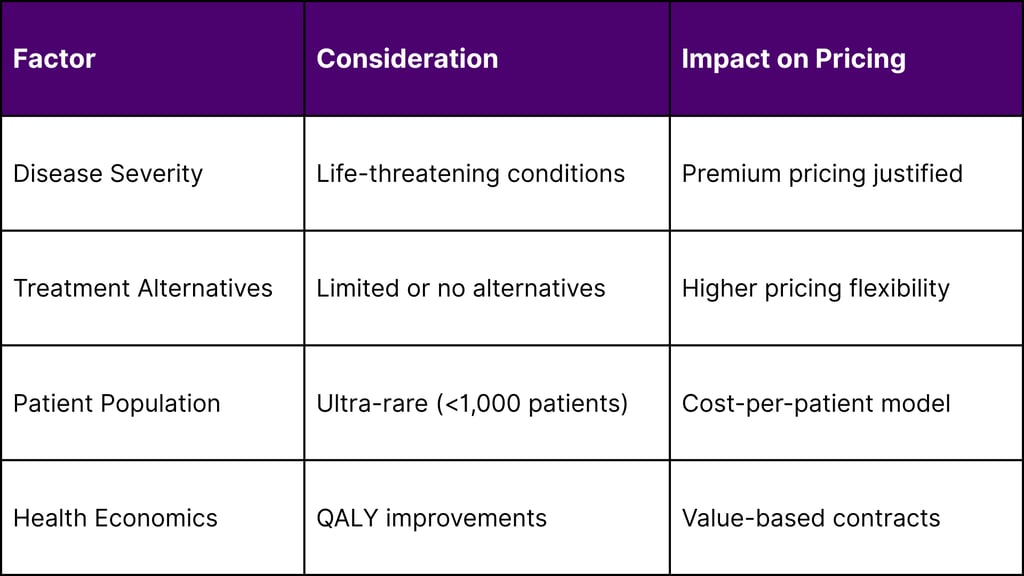
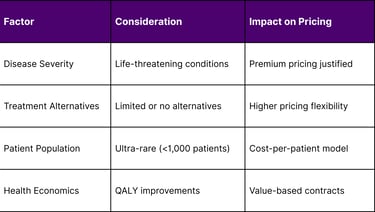
Market Access Considerations
Pricing Strategy Framework:
Regulatory Challenges and Risk Mitigation
Common Pitfalls and Solutions
Challenge 1: Incomplete Natural History Data
Solution: Early investment in natural history studies
Timeline: 2-3 years before pivotal trials
Investment: $2-5 million typical range
Challenge 2: Endpoint Validation
Solution: FDA alignment on endpoints before Phase III
Tools: Biomarker qualification programs
Success Rate: 85% approval rate with pre-agreed endpoints
Challenge 3: Post-Market Commitments
Solution: Proactive confirmatory study design
Timeline: Initiate within 6 months of approval
Compliance Rate: 95% for rare disease confirmatory studies
Future Outlook: Regulatory Evolution
Emerging Trends
Digital Endpoints: Wearable technology and digital biomarkers
Platform Trials: Multi-arm studies for related conditions
Model-Informed Drug Development: Quantitative approaches to dose selection
International Harmonization: Coordinated approvals with EMA and other agencies
Technology Integration
Artificial Intelligence Applications:
Clinical trial optimization
Patient identification and recruitment
Regulatory pathway prediction
Real-world evidence generation
Strategic Recommendations
For Early-Stage Companies
Regulatory Strategy Development: Engage regulatory consultants with rare disease expertise
Biomarker Investment: Allocate 15-20% of R&D budget to biomarker development
FDA Engagement: Schedule pre-IND meetings 12-18 months before first-in-human studies
Partnership Strategy: Consider strategic alliances for regulatory expertise
For Mid-Stage Companies
Accelerated Pathway Assessment: Evaluate eligibility for all expedited programs
Global Strategy: Coordinate FDA and EMA submissions for simultaneous review
Commercial Preparation: Begin market access discussions 2 years before anticipated approval
Risk Management: Develop comprehensive post-market surveillance plans
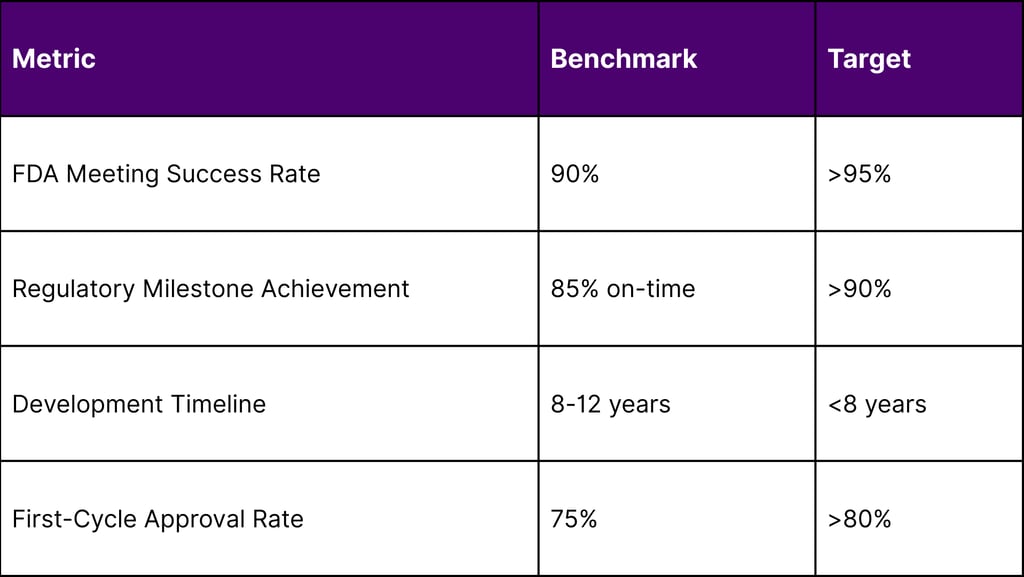
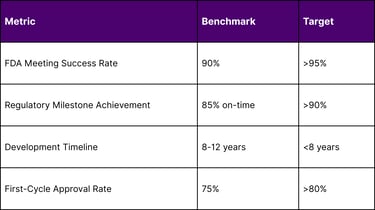
Key Performance Indicators
Conclusion
The regulatory landscape for rare disease therapies offers unprecedented opportunities for companies that understand how to navigate the system effectively. With almost half of all novel medications approved by the U.S. FDA being orphan drugs, the pathway to success is well-established but requires strategic planning and execution.
Success in rare disease drug development depends on three critical factors: early and continuous FDA engagement, robust biomarker strategies, and comprehensive market access planning. Companies that master these elements while leveraging the full spectrum of regulatory tools—from orphan designation to accelerated approval—position themselves for both regulatory success and commercial viability.
The future of rare disease therapeutics is bright, with continued regulatory support, advancing technologies, and growing investment. Organizations that combine scientific innovation with regulatory excellence will continue to transform the lives of patients with rare diseases while building sustainable businesses.
Frequently Asked Questions (FAQ)
Q: When should a company apply for Orphan Drug Designation?
A: Companies can apply for Orphan Drug Designation at any time before submitting their New Drug Application (NDA) or Biologics License Application (BLA). However, applying early (during preclinical or early clinical development) maximizes the benefits, including protocol assistance and potential fee waivers.
Q: What is the success rate for Orphan Drug Designation applications?
A: The FDA grants approximately 85-90% of Orphan Drug Designation requests that meet the basic criteria of affecting fewer than 200,000 patients in the U.S. and demonstrating a plausible hypothesis for treating the condition.
Q: How does accelerated approval differ from standard approval for rare diseases?
A: Accelerated approval allows the FDA to approve drugs based on surrogate or intermediate endpoints that are reasonably likely to predict clinical benefit. Standard approval requires demonstration of clinical benefit on endpoints like survival or irreversible morbidity. Accelerated approval comes with post-market commitments to conduct confirmatory trials.
Q: Can a drug receive both Orphan Drug Designation and accelerated approval?
A: Yes, these designations are complementary. Many rare disease drugs receive orphan designation for the commercial benefits and pursue accelerated approval for faster market entry. This combination is particularly common in oncology and severe rare diseases.
Q: What happens if a company fails to complete post-market commitments under accelerated approval?
A: The FDA has enhanced enforcement mechanisms under the Food and Drug Omnibus Reform Act of 2022, including the ability to withdraw approval more efficiently. Companies should proactively communicate with FDA about study timelines and any challenges in completing confirmatory trials.
Q: How should companies approach international regulatory strategy for rare disease drugs?
A: Companies should coordinate with multiple agencies early in development. The FDA, EMA, and other agencies often accept similar data packages for rare diseases. Consider joint scientific advice procedures and utilize orphan drug designations in multiple jurisdictions simultaneously.
References
U.S. Food and Drug Administration. (2025). Designating an Orphan Product: Drugs and Biological Products. Retrieved from FDA.gov
U.S. Food and Drug Administration. (2025). 9 Things to Know About CDER's Efforts on Rare Diseases. Retrieved from FDA.gov
EveryLife Foundation for Rare Diseases. (2022). FDA Accelerated Approval. Retrieved from everylifefoundation.org
U.S. Food and Drug Administration. (2025). Accelerating Rare disease Cures (ARC) Program. Retrieved from FDA.gov
National Academy of Sciences. (2024). Regulatory Processes for Rare Disease Drugs in the United States and European Union. The National Academies Press.
U.S. Food and Drug Administration. (2024). Prescription Drug User Fee Rates for Fiscal Year 2025. Retrieved from FDA.gov
National Institutes of Health. (2024). Rare Diseases Clinical Research Network. Retrieved from NIH.gov
European Medicines Agency. (2024). Orphan Designation: Overview. Retrieved from EMA.europa.eu
National Organization for Rare Disorders. (2024). Rare Disease Database. Retrieved from rarediseases.org
U.S. Government Accountability Office. (2024). Drug Development: FDA's Expedited Approval Programs. Retrieved from GAO.gov
Navigation
© 2026 FyreIgnis Market Research. All rights reserved.
Legal
info@fyreignismarketresearch.com
India
