
Navigating the Reimbursement Maze for Ultra-Rare Therapies


The landscape of rare disease therapeutics has transformed dramatically over the past four decades. Since the passage of the Orphan Drug Act in 1983, the U.S. Food and Drug Administration (FDA) has approved hundreds of drugs for rare diseases yet the path from regulatory approval to actual patient access remains one of the most complex challenges facing commercial teams today, especially for companies with lean resources.
Navigating the reimbursement maze for ultra-rare therapies requires a precise, multi-pronged market access strategy. For small, agile commercial teams, the pressure is amplified: every payer conversation, every value dossier, and every patient support program must be executed efficiently and with precision. This article breaks down the reimbursement environment, the key barriers, and the strategic levers that lean teams can pull to ensure patients access life-changing therapies.
The Ultra-Rare Disease Landscape: Setting the Stage
The FDA defines a rare disease as one affecting fewer than 200,000 people in the United States. Ultra-rare diseases typically affect far smaller populations sometimes fewer than 10,000 patients nationwide. Understanding the scale of this challenge begins with the numbers.
Key Statistics at a Glance
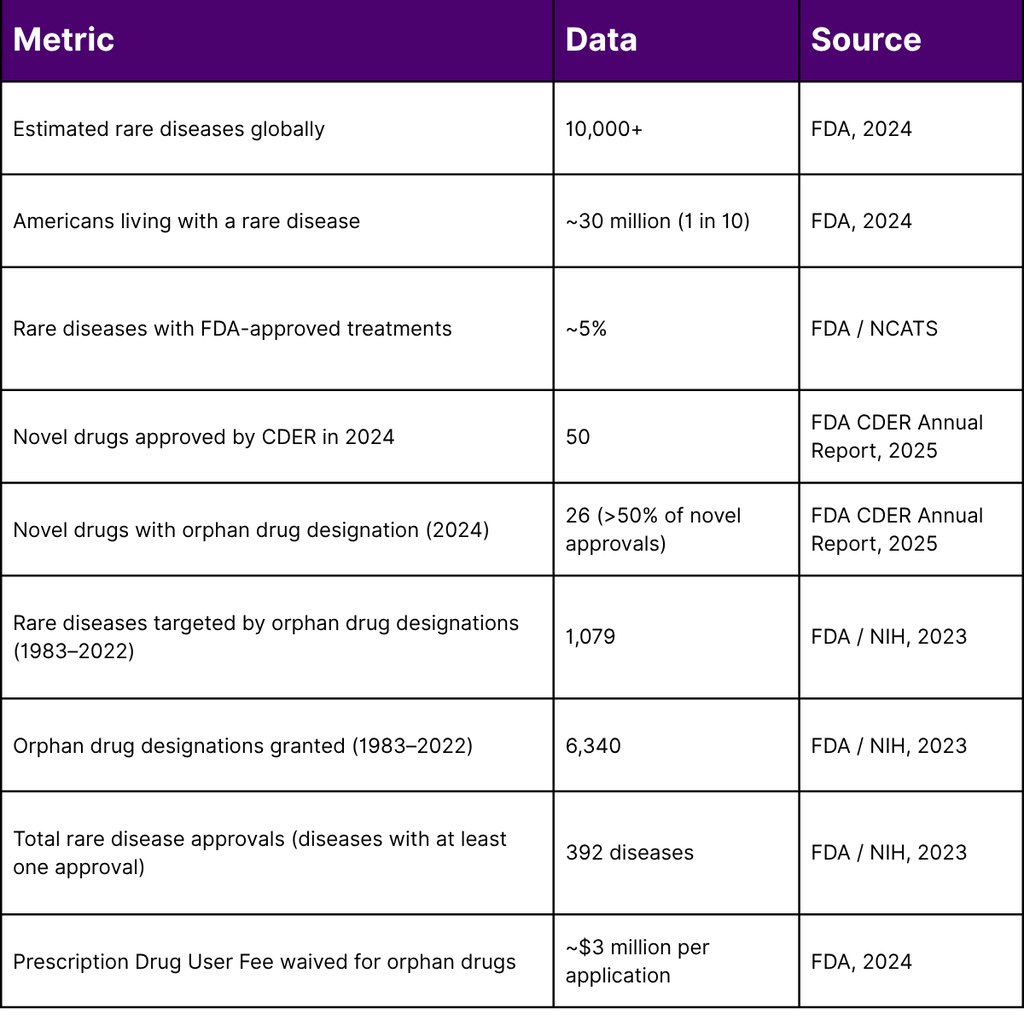
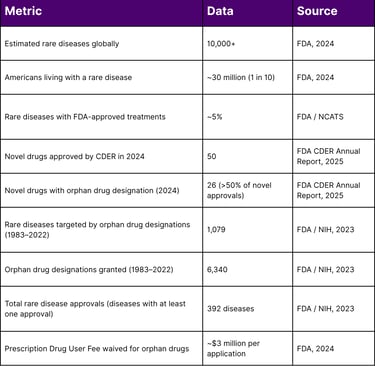
Key Insight: In 2024, therapies with orphan drug designations accounted for more than 50% of all novel drug approvals by CDER including two first-ever treatments for Niemann-Pick Disease Type C and WHIM Syndrome. Yet with only ~5% of rare diseases having any approved treatment, the opportunity and the obligation to improve market access has never been greater.
The Reimbursement Landscape: Why Ultra-Rare Is Different
The Unique Access Barriers
Ultra-rare therapies face a fundamentally different set of market access challenges compared to traditional pharmaceuticals:
1. Limited Clinical Evidence: Randomized controlled trials (RCTs) the gold standard for evidence are difficult or impossible to conduct when a disease affects only a few hundred or thousand patients. According to a 2025 study published via the FDA, orphan drugs approved between 2016 and 2023 were approved based on an average of 1.5 studies per drug, compared to 2.4 studies per drug for non-orphan drugs. Only 34% of orphan drugs were evaluated in RCTs, versus 63% for non-orphan drugs.
2. Small, Dispersed Patient Populations: Patients may be scattered across multiple states or countries, making real-world evidence collection difficult and payer justification complex. As the National Institute of Neurological Disorders and Stroke (NINDS) notes, ultra-rare disease patient populations are "small and dispersed," creating regulatory and funding challenges.
3. High Per-Patient Cost: Ultra-rare therapies particularly gene therapies often command high list prices justified by transformative clinical benefit but difficult for standard payer frameworks to absorb. The indirect and non-medical costs of rare diseases collectively are estimated at $548 billion annually, including $150 billion in absenteeism costs alone.
4. Inconsistent Payer Policies: The U.S. healthcare system is fragmented, comprising thousands of private insurers alongside state Medicaid programs and Medicare. Coverage criteria and formulary decisions vary significantly from plan to plan. Research on neuromuscular disease DMTs found meaningful differences in coverage criteria even among the 17 largest commercial insurers.
5. Medicaid Cross-State Complexity Gene therapies for rare diseases are often administered at a limited number of specialized Centers of Excellence. For Medicaid-enrolled patients, this creates a cross-state access problem: the patient's home state pays but the out-of-state provider must obtain treatment authorization separately a process that can introduce costly delays.
The Regulatory Framework: Your Strongest Market Access Ally
Understanding and leveraging FDA regulatory tools is essential for lean commercial teams making the case to payers.
Orphan Drug Act Incentives That Matter for Payer Negotiations
The Orphan Drug Act (enacted 1983) provides a suite of incentives that directly strengthen your market access position:
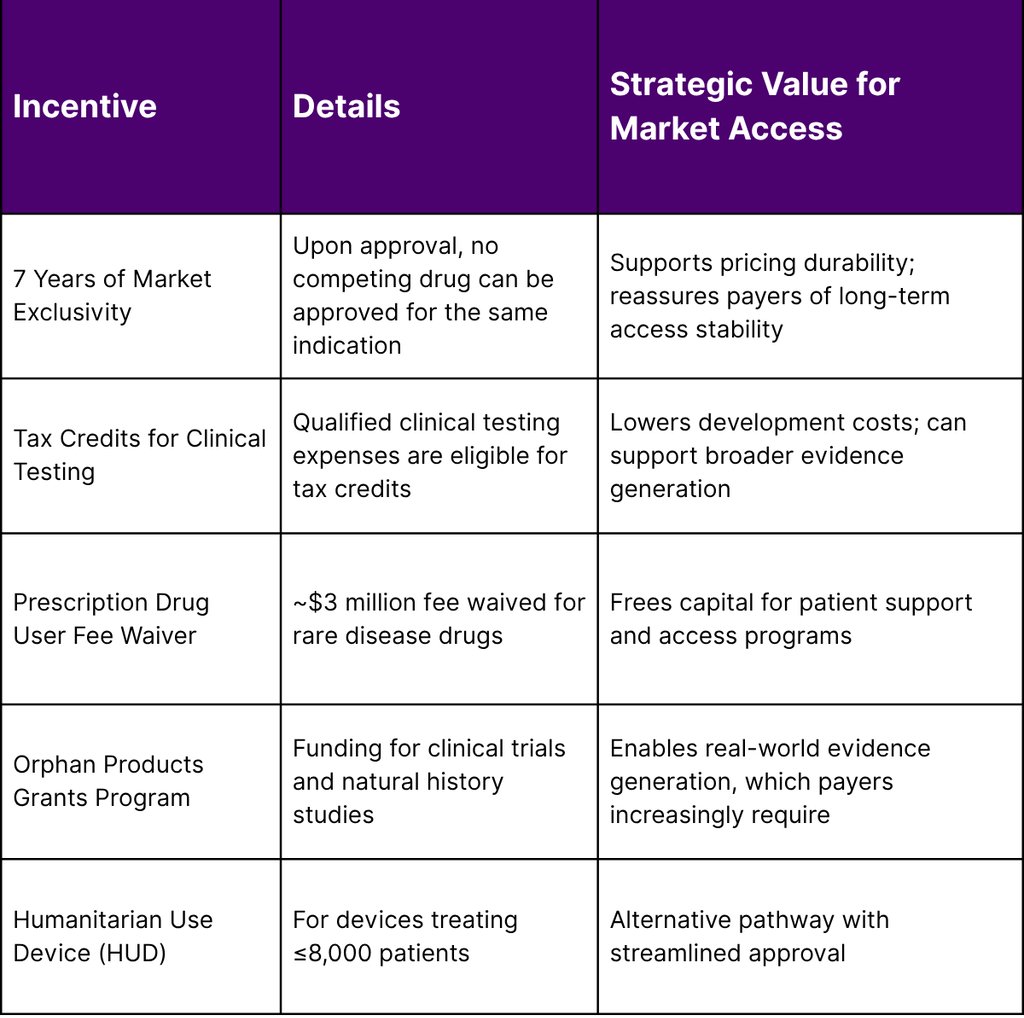
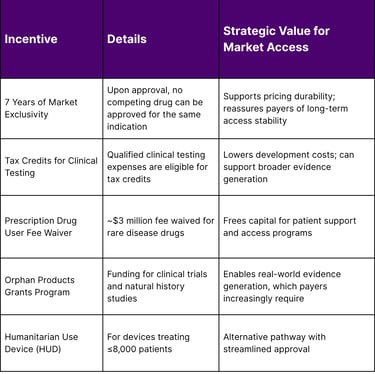
Strategic Tip for Lean Teams: When building payer value dossiers, explicitly highlight your orphan drug designation and any expedited FDA pathways (Breakthrough Therapy, Accelerated Approval, Priority Review). In 2024, CDER used expedited pathways for the majority of rare disease approvals a strong signal of unmet medical need that can anchor payer value conversations.
Market Access Strategies for Lean Commercial Teams
1. Build the Value Story Early — Before Approval
Payer engagement should not begin at launch. Lean teams must begin building evidence packages during Phase II or III clinical development:
Map the payer landscape early: Identify which payer segments (commercial, Medicaid, Medicare Part B vs. D) are most relevant given your patient population's demographics.
Develop a robust natural history evidence package: Since clinical trials in ultra-rare diseases are small, payers will increasingly require real-world and natural history data. The NIH's Rare Diseases Clinical Research Network (RDCRN) supports natural history studies that can serve as valuable comparator data.
Quantify the burden of disease in economic terms: Total indirect and non-medical costs of rare diseases are estimated at $548 billion annually. Frame your therapy's value in terms of avoided hospitalizations, caregiver burden reduction, and productivity gains.
2. Master the Medicaid Strategy
Medicaid is a critical payer for rare disease patients, particularly children. Key strategic points:
Mandatory coverage of FDA-approved drugs applies: Under the Medicaid Drug Rebate Program (MDRP), states are generally required to cover FDA-approved drugs when manufacturers have a signed rebate agreement with the Secretary of HHS. This is a foundational protection for patient access.
Rebate obligations must be planned for: Innovator drugs are subject to a minimum rebate of 23.1% of average manufacturer price (AMP) under the ACA, with exceptions for drugs with only pediatric indications (17.1% of AMP).
Advocate for cross-state Medicaid solutions: Federal legislation such as the Accelerating Kids' Access to Care Act addresses out-of-state provider enrollment barriers. Lean teams should monitor and support such legislative developments.
3. Leverage Patient Assistance and Support Programs Strategically
For ultra-rare therapies administered at Centers of Excellence, travel and lodging burdens are real barriers. The FDA has established precedents for federal safe harbors that allow manufacturers to offer travel and lodging support a model first applied with Kymriah (for CAR-T). Lean commercial teams should:
Work with legal counsel to structure compliant patient support programs.
Partner with rare disease patient advocacy organizations, which often serve as trusted intermediaries for access support.
Build bridge programs for patients awaiting payer decisions.
4. Develop Real-World Evidence (RWE) to Support Formulary Decisions
Private payers increasingly use real-world evidence to inform formulary and coverage decisions, especially when head-to-head clinical trials are unavailable a near-constant reality in ultra-rare diseases. Lean teams should:
Establish patient registries from launch (the NIH provides best-practice guidance for patient registries through NCATS).
Partner with academic medical centers and Centers of Excellence to collect longitudinal outcome data.
Use biomarker and surrogate endpoint data where validated, consistent with FDA's approach to accelerated approval pathways.
5. Design for Outcomes-Based Agreements
Given the high per-patient cost of ultra-rare therapies and limited clinical evidence, outcomes-based or managed entry agreements (MEAs) are increasingly viable tools. These may include:
Coverage with evidence development (CED): Payer covers the therapy in exchange for real-world data collection post-launch.
Milestone-based payment arrangements: Particularly relevant for one-time gene therapies.
Stop-loss reinsurance partnerships: Payers can participate in stop-loss reinsurance to protect against catastrophic individual claims a model worth educating payers on during early engagement.
A Framework for the Lean Commercial Team: Prioritization Matrix
The following framework helps small teams allocate limited resources across market access priorities:
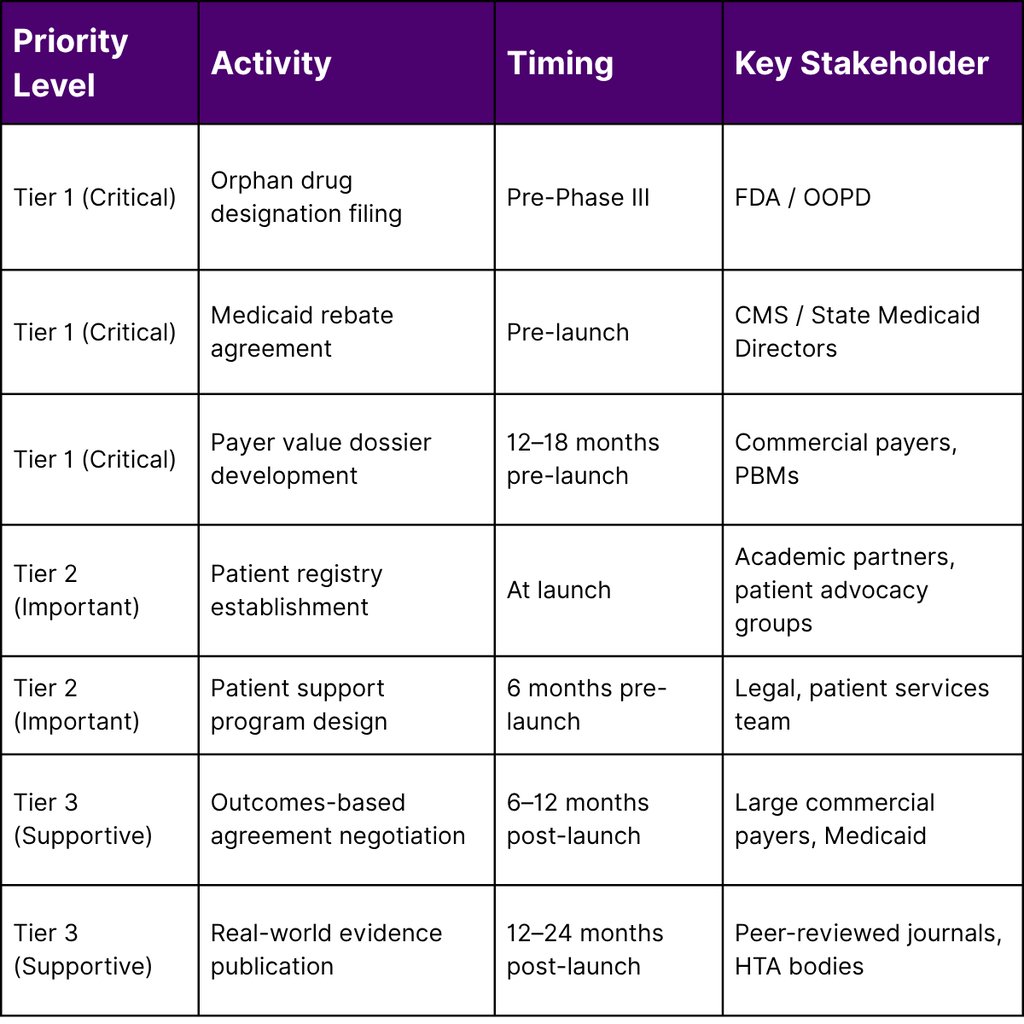
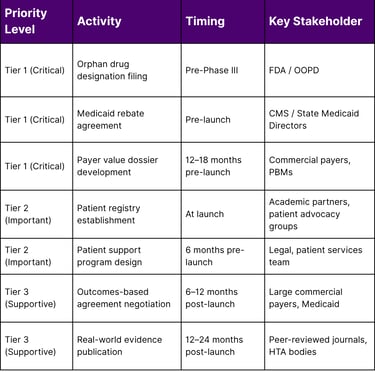
The Gene Therapy Reimbursement Challenge: A Special Case
Gene and cell therapies represent perhaps the most complex reimbursement challenge within the ultra-rare space. As of 2025, the FDA has approved a growing number of gene therapies, yet commercial sustainability remains fragile. Pfizer discontinued its FDA-approved gene therapy Beqvez for hemophilia B less than a year after approval in early 2025 due to limited patient uptake illustrating how regulatory approval alone does not ensure patient access or commercial viability.
For gene therapy commercial teams specifically:
The one-time administration model disrupts traditional annual rebate and formulary structures.
Payment spreading mechanisms (annuity-based payments, installment agreements) require novel contracting vehicles.
Multi-state Medicaid coordination requires proactive federal and state policy engagement.
The NINDS URGenT network and NCATS' Bespoke Gene Therapy Consortium (BGTC) are developing platform standards to accelerate gene therapy development a signal that collaborative infrastructure is being built that lean commercial teams can leverage.
Conclusion
The reimbursement environment for ultra-rare therapies is complex, fragmented, and evolving but it is navigable. Lean commercial teams that begin payer engagement early, build robust real-world evidence programs, leverage FDA regulatory incentives strategically, and design innovative access solutions are best positioned to ensure that patients reach the therapies they need.
With over 10,000 identified rare diseases and only ~5% having approved treatments, the market access imperative is not just commercial it is a patient-centered mission. At Fyreignis Market Research, we help commercial teams decode the payer landscape, develop evidence strategies, and accelerate access for the patients who need it most.
Frequently Asked Questions (FAQ)
Q1: What is the difference between a rare disease and an ultra-rare disease? The FDA defines a rare disease as one affecting fewer than 200,000 people in the United States. "Ultra-rare" is a subset not formally defined in U.S. law typically used to describe conditions affecting far fewer patients, sometimes fewer than 1,000 to 10,000 individuals. The Humanitarian Use Device (HUD) designation, for example, covers devices for conditions affecting 8,000 or fewer U.S. patients.
Q2: Are Medicaid programs required to cover FDA-approved orphan drugs? Generally, yes. Under the Medicaid Drug Rebate Program (MDRP), states that receive federal Medicaid funding are required to cover FDA-approved outpatient prescription drugs when the manufacturer has a signed rebate agreement with HHS. However, prior authorization, step therapy, and other utilization management requirements can still create access barriers in practice.
Q3: What is the Orphan Drug Act's market exclusivity provision? The Orphan Drug Act provides seven years of market exclusivity upon FDA approval for drugs with orphan drug designation. This means the FDA will not approve the same drug for the same approved orphan indication during the exclusivity period, though it can approve other drugs for that disease and other indications for the same drug.
Q4: How can a lean commercial team build real-world evidence with limited resources? Key strategies include: partnering with academic medical centers and Centers of Excellence already treating your patient population; establishing patient registries with NIH best-practice guidance (NCATS provides free resources); leveraging natural history data from NIH-funded programs such as the Rare Diseases Clinical Research Network; and designing data collection into patient support programs from day one.
Q5: What are outcomes-based agreements and are they viable for ultra-rare therapies? Outcomes-based or managed entry agreements (MEAs) link payment to real-world clinical performance. For ultra-rare therapies, these may include coverage with evidence development (CED) arrangements or milestone-based payments for gene therapies. While administratively complex, they can be a viable path to formulary access when clinical evidence at launch is limited a common scenario in ultra-rare diseases.
Q6: What FDA expedited pathways are most relevant for ultra-rare therapies? Key expedited pathways include: Breakthrough Therapy Designation, Accelerated Approval (using surrogate endpoints), Priority Review (six-month vs. standard ten-month review), and Fast Track designation. In 2024, CDER used a combination of these tools for the majority of rare disease approvals, and demonstrating these designations to payers strengthens the unmet need argument in coverage discussions.
Q7: What role do patient advocacy organizations play in market access? Patient advocacy organizations (PAOs) are critical partners for rare disease market access. They can support payer education about disease burden, assist in patient identification and registry enrollment, advocate for coverage policies at the state and federal level, and provide trusted patient navigation services. For lean teams, PAO partnerships can extend commercial reach significantly.
References
U.S. Food and Drug Administration. (2024). Rare diseases at FDA.
U.S. Food and Drug Administration, Center for Drug Evaluation and Research. (2025). Advancing health through innovation: New drug therapy approvals 2024.
U.S. Food and Drug Administration. (2024). Rare disease drug approvals.
U.S. Food and Drug Administration. (2024). Designating an orphan product: Drugs and biological products.
U.S. Food and Drug Administration. (2024). Orphan Drug Act — relevant excerpts.
U.S. Food and Drug Administration. (2024). Drug development for very rare diseases.
U.S. Food and Drug Administration. (2024). Medical products for rare diseases and conditions.
U.S. Food and Drug Administration. (2024). FDA unveils plan to eliminate orphan designation backlog.
National Center for Advancing Translational Sciences (NCATS). (2024). Our impact on rare diseases. National Institutes of Health.
National Center for Advancing Translational Sciences (NCATS). (2025). Additional rare diseases research and initiatives. National Institutes of Health.
National Center for Advancing Translational Sciences (NCATS). (2024). Resources for rare disease patients and advocates. National Institutes of Health.
National Institute of Neurological Disorders and Stroke (NINDS). (2021). NINDS launches URGenT: A network to accelerate the development of treatments for ultra-rare neurological diseases. National Institutes of Health.
National Institutes of Health, National Center for Advancing Translational Sciences. (2025). PAR-25-450: Clinical trial readiness for rare diseases, disorders, and syndromes (R21 Clinical Trial Not Allowed).
Fermaglich, L. J., & Miller, K. L. (2023). A comprehensive study of the rare diseases and conditions targeted by orphan drug designations and approvals over the forty years of the Orphan Drug Act. Orphanet Journal of Rare Diseases, published June 23, 2023. [Note: Authors affiliated with FDA Office of Orphan Products Development]
Koong, A. J., Irvin, V. L., Narayan, A., Song, S., & Kaplan, R. M. (2025). Evidence available and used by the Food and Drug Administration for the approval of orphan and nonorphan drugs. Health Affairs Scholar, published March 18, 2025.
Berry, D., Hickey, C., Kahlman, L., Long, J., Markus, C., & McCombs, C. K. (2025). Ensuring patient access to gene therapies for rare diseases: Navigating reimbursement and coverage challenges. Molecular Therapy: Methods & Clinical Development, 33(1), 101403.
Centers for Medicare & Medicaid Services. Medicaid Drug Rebate Program. Referenced in: National Academies of Sciences. Coverage and reimbursement: Incentives and disincentives for product development.
Navigation
© 2026 FyreIgnis Market Research. All rights reserved.
Legal
info@fyreignismarketresearch.com
India
