
Patient-Centricity in Oncology


Oncology drug development is one of the most competitive arenas in life sciences. In 2024, the FDA's Office of Oncologic Diseases approved 17 novel drugs new molecular entities or biologics covering gynecologic, genitourinary, lung, gastrointestinal, breast, thyroid, and hematologic malignancies. Beyond CDER, the FDA's Center for Biologics Evaluation and Research approved two new cellular therapy products for cancer in the same year. Altogether, the FDA facilitated 76 oncology device authorizations in 2024 alone.
With this level of activity, the question for any drug developer is not just whether a therapy works, but whether it works in ways that matter most to patients. The commercial difference between a "me-too" approval and a market-defining franchise increasingly hinges on one thing: how deeply and how early patient insights were embedded into clinical design.
This article examines why involving patient advocacy groups early in the clinical design phase leads to better regulatory, commercial, and scientific outcomes and provides a framework for doing so systematically.
The Oncology Landscape: Scale and Stakes
To understand why patient-centricity in oncology is a strategic imperative, one must first understand the scale of the patient population and the urgency driving drug development.
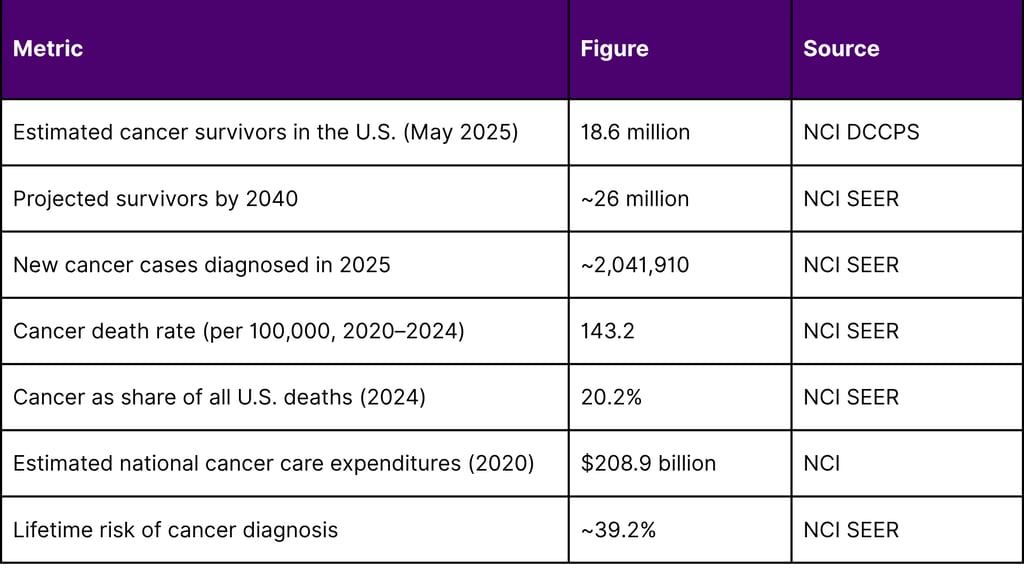
As of May 2025, there are an estimated 18.6 million cancer survivors in the United States, representing approximately 5.4% of the total population. From 2022 to 2040, the number of people who have lived five or more years after their cancer diagnosis is projected to increase approximately 53%, reaching 19.2 million. In 2025, an estimated 2,041,910 new cancer cases were diagnosed in the United States, and 618,120 people died from the disease.
The financial scale is equally significant. Estimated national expenditures for cancer care in the United States in 2020 were $208.9 billion, with costs expected to increase as the population ages.
Table 1: Key U.S. Oncology Statistics (NCI SEER / FDA)
Yet despite the scale of the patient population, clinical trial enrollment remains alarmingly low. The overall estimated patient participation rate in cancer treatment trials is 7.1%, with enrollment reaching 21.6% only at NCI-designated comprehensive cancer centers, and dropping to 4.1% at community programs. Historically, only approximately 3% of adult cancer patients participate in clinical trials.
This enrollment gap is not merely a logistical challenge it is a signal of a design problem. Trials that do not reflect what patients consider burdensome, tolerable, or meaningful will fail to enroll, fail to retain, and ultimately fail to generate evidence that is commercially persuasive.
The Regulatory Mandate for Patient Centricity
The shift toward patient-centricity in oncology is not merely an industry trend. It is now federal regulatory policy.
The 21st Century Cures Act (2016)
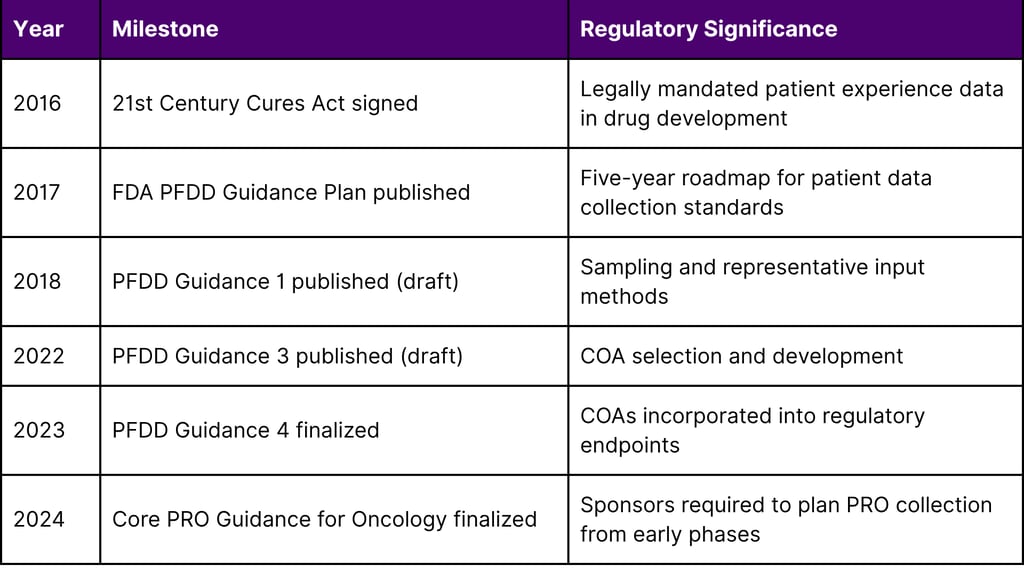
On December 13, 2016, the 21st Century Cures Act was signed into law. Title III, Section 3001 defines patient experience data as data "collected by any persons and intended to provide information about patients' experiences with a disease or condition, including the impact physical and psychosocial of such disease or condition, or a related therapy, on patients' lives, and patient preferences with respect to treatment."
Section 3002 directed the FDA to issue a series of guidance documents on how to collect, measure, and use patient experience data in drug development. Section 3001 additionally requires the FDA to make public a brief statement on whether and how patient experience data was used in any drug approval decision.
The FDA's Four-Part PFDD Guidance Series
In response to the 21st Century Cures Act and commitments under PDUFA VI, the FDA developed a series of four methodological Patient-Focused Drug Development (PFDD) guidance documents. This series is intended to facilitate the advancement and use of systematic approaches to collect and use robust and meaningful patient and caregiver input that can better inform medical product development and regulatory decision-making.
Guidance 1 addresses who to collect input from, including sampling methods and target population definition
Guidance 2 discusses methods for eliciting information from patients about what aspects of their disease and treatment experience matter most
Guidance 3 discusses approaches to selecting, modifying, developing, and evaluating clinical outcome assessments (COAs) to measure outcomes of importance to patients
Guidance 4 addresses how to incorporate clinical outcome assessments into endpoints for regulatory decision-making
The FDA's October 2024 Core PRO Guidance for Oncology
In October 2024, the FDA's Oncology Center of Excellence (OCE) finalized the "Core Patient-Reported Outcomes in Cancer Clinical Trials" guidance described by the agency as "a key milestone for the OCE PFDD program" and "a distillation of many publications and years of regulatory work."
This guidance provides recommendations for collecting a core set of patient-reported clinical outcomes in cancer clinical trials and related considerations for instrument selection and trial design. The FDA recommends collecting and analyzing the following core PROs: disease-related symptoms, symptomatic adverse events, an overall side effect impact summary measure, and physical and role function.
Critically, the guidance states that assessment of key symptoms is feasible at all stages of drug development, including incorporation of PROs in early phase drug development.
This sentence carries significant strategic weight: the FDA is not merely asking sponsors to collect PRO data in Phase III. It is signaling that patient-centered evidence generated in Phase I and Phase II trials will carry regulatory and labeling implications.
Table 2: FDA Oncology PFDD Regulatory Milestones (2016–2024)
Why Early Advocacy Group Involvement Drives Better Commercial Outcomes
Patient advocacy groups (PAGs) occupy a unique position in the oncology ecosystem. They are simultaneously the voice of the end user, the custodians of patient community trust, and increasingly sophisticated scientific stakeholders with deep knowledge of disease biology, unmet needs, and quality-of-life priorities.
When pharmaceutical sponsors engage PAGs only at the end of a trial during recruitment campaigns or post-approval access programs they forfeit significant strategic value. Early engagement, by contrast, creates compounding returns across six dimensions.
1. Endpoint Selection That Survives Regulatory Scrutiny
The FDA's Core PRO Guidance defines what oncology patients care about: disease-related symptoms, treatment side effects, and functional capacity. But within these categories, the specific symptoms and the specific function domains that matter most vary by cancer type, treatment modality, and patient population.
A lung cancer patient undergoing first-line immunotherapy has fundamentally different symptom priorities than a patient with advanced hematologic malignancy receiving CAR-T cell therapy. Advocacy groups with embedded experience in specific tumor types can inform which PRO instruments are fit for purpose, which symptom items are most burdensome, and which functional domains represent meaningful clinical change.
Sponsors who get endpoint selection wrong face a painful outcome: efficacy signals that cannot be translated into labeling language because the PRO measures were not validated, sensitive, or meaningful enough to satisfy regulators. Getting endpoint selection right from the Phase I/II design stage is therefore a direct regulatory risk mitigation strategy.
2. Protocol Design That Patients Can Tolerate
The early and ongoing involvement of patient advocacy communities in designing and conducting clinical trials has resulted in some of the most robust improvements in cancer treatment to date, according to data presented at an Institute of Medicine public session on cancer clinical trials.
PAGs are uniquely positioned to flag protocol elements that create disproportionate burden: excessive biopsy requirements, frequent clinic visit schedules incompatible with functional limitations, eligibility criteria that exclude patients with comorbidities prevalent in the real-world patient population, and assessment schedules that overwhelm patients already experiencing disease burden.
Incorporating these insights into protocol design reduces dropout rates, increases the representativeness of the enrolled population, and improves the quality of the safety and efficacy data generated. All three of these factors directly affect the strength of the regulatory submission.
3. Recruitment Speed and Enrollment Quality
As established above, only 7.1% of cancer patients participate in clinical trials overall. At community programs which are where the majority of cancer patients receive care — the rate falls to 4.1%. Patient advocacy groups are trusted intermediaries between the scientific and commercial world and the patient community. Sponsors who cultivate relationships with advocacy organizations before a trial begins have access to engaged, informed patient communities who understand the protocol, trust the sponsor, and can refer eligible candidates through established networks.
This is not a marginal benefit. Enrollment delays are one of the most significant drivers of clinical development cost and timeline. Faster enrollment through advocacy-supported recruitment compounds commercial advantage.
4. Unmet Need Documentation That Supports Labeling
The FDA's PFDD guidance framework enables sponsors to submit patient experience data as part of a drug application. When this data is rich, longitudinal, and validated including qualitative interviews with advocacy group members documenting the lived experience of the disease it provides the regulatory foundation for differentiated labeling language.
Labeling that captures specific patient-reported symptoms, functional limitations, and quality-of-life improvements is not merely a scientific achievement. It is a commercial asset. Payers, prescribers, and health technology assessment bodies use labeling language to make formulary and reimbursement decisions. Sponsors whose labels reflect meaningful patient benefit documented with patient-generated evidence have a structural advantage in market access negotiations.
5. Alignment with FDA Priority Designations
In 2024, the FDA approved 33 of 50 new drugs using one or more expedited approval programs fast track, breakthrough therapy, priority review, or accelerated approval. Breakthrough therapy designation, in particular, requires preliminary clinical evidence that a drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint.
What counts as clinically significant? Increasingly, the answer is shaped by patient experience data. Sponsors who have invested in patient insights who know which symptoms are most burdensome, which functional impairments are most clinically meaningful, and which treatment effects are most valued are better positioned to design early clinical studies that generate the kind of evidence needed to qualify for breakthrough designation.
Additionally, in 2024, 26 of 50 novel drug approvals had previously received orphan drug designation. PAGs focused on rare cancers often serve as the primary scientific and regulatory navigation partners for small and mid-sized biotechs developing therapies for these indications.
6. Trust-Building That Creates Long-Term Commercial Relationships
Beyond the immediate clinical development cycle, early and authentic engagement with PAGs creates durable trust. Advocacy organizations shape the treatment narrative in patient communities, influence physician education, and can advocate at payer and policy levels on behalf of therapies they trust.
Conversely, sponsors who are perceived as extractive consulting patients only for recruitment purposes, not incorporating advocacy input into trial design generate reputational risks that compound over time, particularly in rare disease and pediatric oncology communities where patient networks are tightly connected and long memories are the norm.
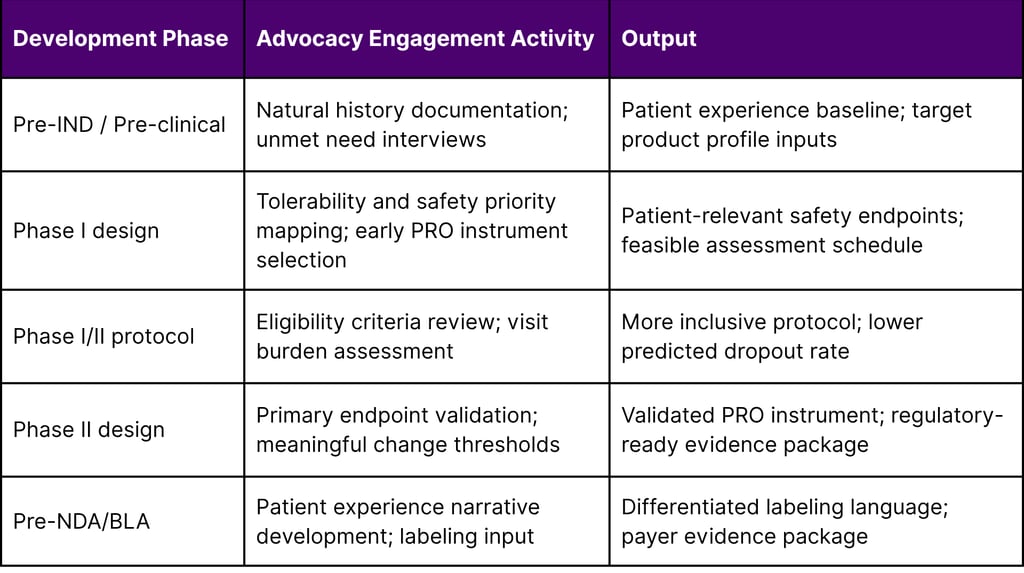
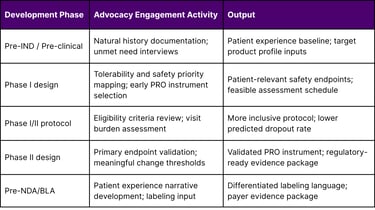
A Framework for Early Advocacy Group Engagement in Oncology Clinical Design
The following framework describes a systematic approach to embedding patient advocacy insights from the earliest phases of clinical development.
Table 3: Patient Advocacy Engagement Framework by Development Phase
Expedited Programs and Patient Centricity: A Virtuous Cycle
The relationship between patient-centered clinical design and FDA expedited pathways is not coincidental it is structural. The FDA's breakthrough therapy designation, fast track designation, and accelerated approval pathway all reward sponsors who can demonstrate understanding of unmet patient need and design studies that generate clinically meaningful evidence efficiently.
In the FDA's own language, Breakthrough Therapy designation is "a process designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint." Both the seriousness of the condition and the clinical significance of the endpoint are ultimately grounded in the patient experience.
In 2024, the FDA's OCE facilitated 23 product approvals through Project Orbis a collaboration with international regulatory partners including five new products and 18 indication extensions. In 2024, the OCE also processed 761 single-patient expanded access applications through Project Facilitate. Both programs reflect the FDA's commitment to getting promising therapies to patients efficiently, and both are accelerated by sponsors who bring coherent, patient-grounded evidence packages.
Table 4: FDA Oncology Expedited Programs and Patient-Centricity Linkage
Conclusion
The oncology drug development landscape is simultaneously more productive and more competitive than at any point in history. In 2024 alone, the FDA approved 17 novel oncology drugs, processed nearly 800 expanded access applications, and finalized landmark guidance embedding patient-reported outcomes at the center of cancer trial design. With an estimated 18.6 million cancer survivors in the United States and projections pointing to 26 million by 2040, the patient population driving this market continues to grow.
In this environment, patient-centricity is no longer a differentiator it is a prerequisite. The sponsors who will build durable competitive moats are those who engage patient advocacy groups at the protocol design stage, not the recruitment stage. They are the ones who understand, before the first patient is enrolled, which symptoms matter most, which functional losses are most burdensome, and which treatment outcomes would be genuinely meaningful.
That understanding is not just good science. It is a commercially decisive asset that shapes regulatory pathways, enrollment timelines, labeling negotiations, and long-term market access.
At Fyreignis Market Research, we help life sciences organizations translate patient advocacy intelligence into clinical design strategy.
Frequently Asked Questions
Q1: What exactly is patient experience data under U.S. law?
Under Title III, Section 3001 of the 21st Century Cures Act, as amended by Section 605 of the FDA Reauthorization Act of 2017 (FDARA), patient experience data includes data collected by any persons and intended to provide information about patients' experiences with a disease or condition. This encompasses the physical and psychosocial impact of disease and related therapies, as well as patient preferences with respect to treatment.
Q2: When did the FDA first require patient experience data to be disclosed in drug applications?
The 21st Century Cures Act (signed December 13, 2016) requires FDA to make public a brief statement on whether and how patient experience data was used in the review of any drug or biologic marketing application. A new subsection called "Patient Experience Data" was subsequently added to drug and biologic review documents as a result.
Q3: What are core PROs in oncology, and why did the FDA finalize guidance for them in 2024?
Core PROs in oncology are patient-reported outcomes that the FDA recommends sponsors collect across all registrational cancer trials: disease-related symptoms, symptomatic adverse events, overall side effect impact, and physical and role function. The guidance was finalized in October 2024 by the FDA's Oncology Center of Excellence as a distillation of over a decade of regulatory science work. The finalization signals that these measures are now expected elements of oncology trial design.
Q4: How do patient advocacy groups differ from patient advisory boards?
Patient advocacy groups (PAGs) are independent organizations formed by and for patients and caregivers around specific disease areas. They represent patient communities at the legislative, regulatory, scientific, and commercial levels. Patient advisory boards are typically sponsor-convened groups of patients engaged for specific consultation purposes. For regulatory credibility and authentic community trust, engagement with established independent PAGs carries more weight than internal advisory board activities.
Q5: Can small biotechs with limited resources implement a patient advocacy engagement strategy?
Yes. The FDA's PFDD guidance framework, its OCE Rare Cancers Program, and the NIH's research databases (NIH RePORTER, ClinicalTrials.gov) all provide publicly accessible infrastructure that small sponsors can use to identify advocacy groups, understand unmet needs, and access patient experience data. The FDA's Project Catalyst also welcomes questions regarding oncology drug development plans that are premature for a formal pre-IND submission, providing a low-barrier entry point for early dialogue.
Q6: Does patient-centered clinical design affect the FDA's evaluation of PRO data in drug labeling?
Yes. The FDA's PFDD Guidance 4 (finalized 2023) addresses specifically how clinical outcome assessments are incorporated into endpoints for regulatory decision-making. Sponsors whose PRO measures are fit-for-purpose, validated, and designed around meaningful patient anchors are more likely to have PRO data reflected in their approved product labeling.
References
U.S. Food and Drug Administration. (2024). 2024 OCE annual report. FDA Oncology Center of Excellence.
U.S. Food and Drug Administration. (2024). Core patient-reported outcomes in cancer clinical trials: Guidance for industry. FDA Center for Drug Evaluation and Research; Center for Biologics Evaluation and Research.
U.S. Food and Drug Administration. (2024). Oncology regulatory review 2024. FDA Oncology Center of Excellence.
U.S. Food and Drug Administration. (2024). Ongoing clinical oncology projects 2024. FDA Oncology Center of Excellence.
U.S. Food and Drug Administration. (2024). CDER brings many safe and effective therapies to patients and consumers in 2024.
U.S. Food and Drug Administration. (2023). Patient-focused drug development: Incorporating clinical outcome assessments into endpoints for regulatory decision-making (Guidance 4).
U.S. Food and Drug Administration. (2017). FDA patient-focused drug development guidance series.
U.S. Food and Drug Administration. (2016). 21st Century Cures Act: Patient experience data requirements.
U.S. Food and Drug Administration. (n.d.). Breakthrough therapy designation.
National Cancer Institute. (2025). Cancer statistics. NCI SEER Program.
National Cancer Institute, Division of Cancer Control and Population Sciences. (2025). Statistics and graphs: Cancer survivorship.
National Cancer Institute. (2025). Annual report to the nation on the status of cancer.
National Cancer Institute, SEER Program. (2025). Cancer stat facts: Cancer of any site.
Unger, J. M., et al. (2024). National estimates of the participation of patients with cancer in clinical research studies based on Commission on Cancer Accreditation data. Journal of Clinical Oncology, 42(18), 2139–2148.
National Academies of Sciences, Engineering, and Medicine. (2010). A national cancer clinical trials system for the 21st century: Reinvigorating the NCI cooperative group program. National Academies Press.


Navigation
© 2026 FyreIgnis Market Research. All rights reserved.
Legal
info@fyreignismarketresearch.com
India
