
Personalized Gene‑Editing Breakthrough: CPS1 Deficiency


A Groundbreaking Medical Achievement Transforms Rare Disease Treatment
In a historic medical milestone that could revolutionize personalized medicine, researchers have successfully treated an infant with CPS1 deficiency using the world's first customized CRISPR gene-editing therapy. This marks the first time a tailored gene therapy has been safely delivered to a human patient, rewriting the infant's DNA directly inside liver cells.
Understanding CPS1 Deficiency: A Rare but Devastating Condition
Carbamoyl phosphate synthetase 1 (CPS1) deficiency is one of the most severe forms of urea cycle disorders, a group of inherited conditions that affect the body's ability to remove ammonia from the blood. Ammonia, which is formed when proteins are broken down in the body, is toxic if the levels become too high. The nervous system is especially sensitive to the effects of excess ammonia.
Epidemiology and Clinical Impact
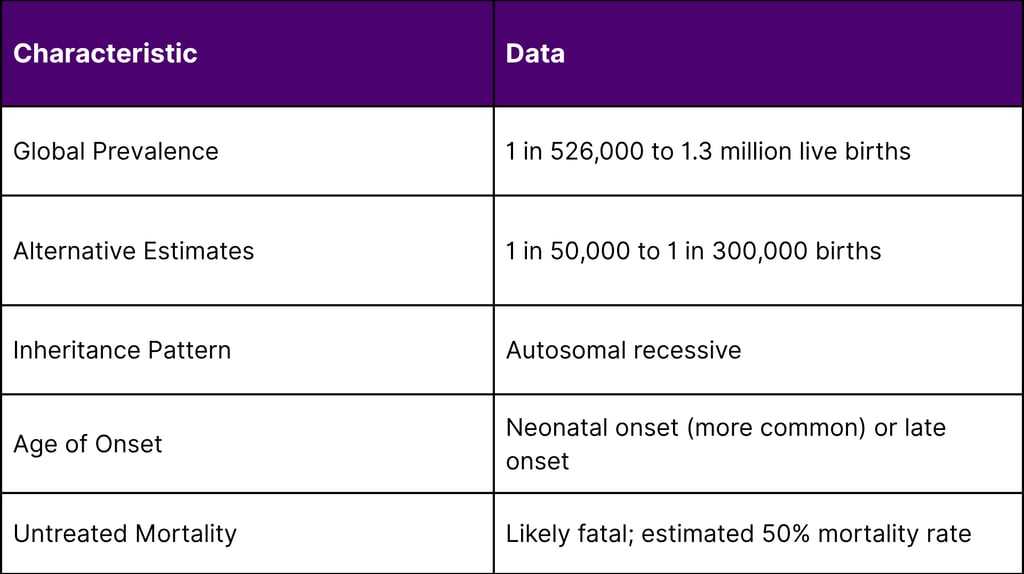
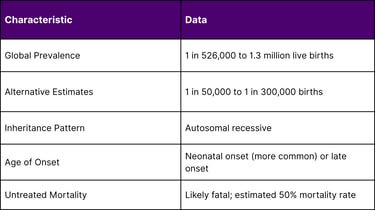
The rarity of CPS1 deficiency makes it particularly challenging to study and treat:
Clinical Presentation
In the neonatal-onset form of CPS1 deficiency, patients are usually healthy at birth but after few days they begin to manifest with lethargy and unwillingness to feed. The condition rapidly progresses to life-threatening hyperammonemia, requiring immediate intervention to prevent neurological damage or death.
The Revolutionary Treatment Approach
Patient Profile: KJ's Journey
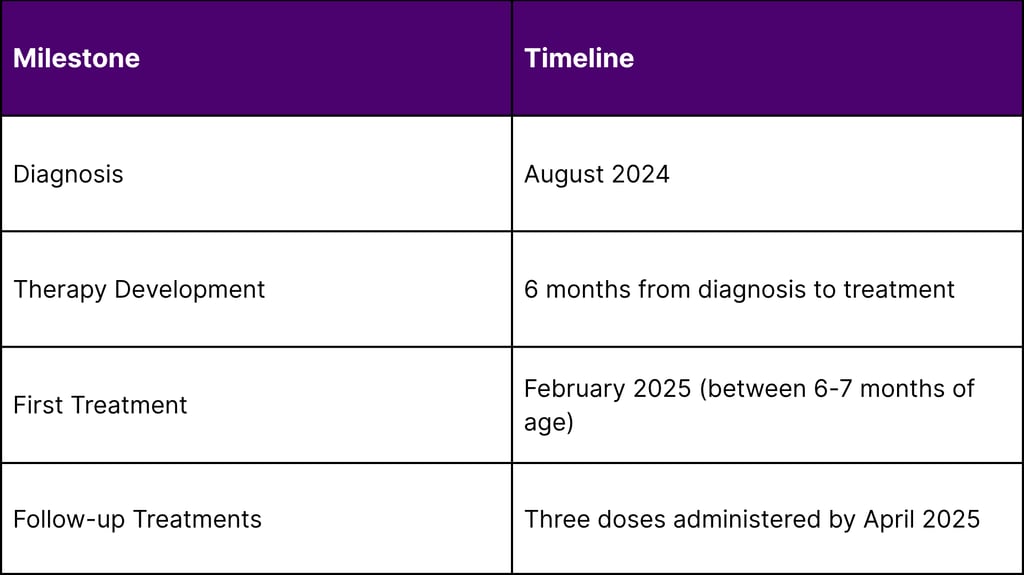
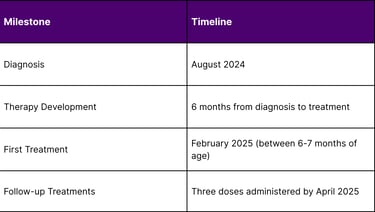
The groundbreaking treatment was administered to an infant known as "KJ," who was diagnosed with severe CPS1 deficiency shortly after birth in August 2024. The infant was diagnosed soon after birth with severe carbamoyl phosphate synthetase 1 deficiency and became the ideal candidate for this experimental therapy due to the severity of his condition.
Treatment Timeline and Methodology
The remarkable speed of development and implementation demonstrates the potential of personalized medicine:
CRISPR Base Editing Technology
The treatment utilized advanced base editing technology, a refined form of CRISPR gene editing that allows for precise single-nucleotide changes without creating double-strand breaks in DNA. Base editors can correct disease-causing genetic variants, making them particularly suitable for addressing point mutations that cause genetic disorders like CPS1 deficiency.
Treatment Outcomes and Clinical Results
Immediate Safety Profile
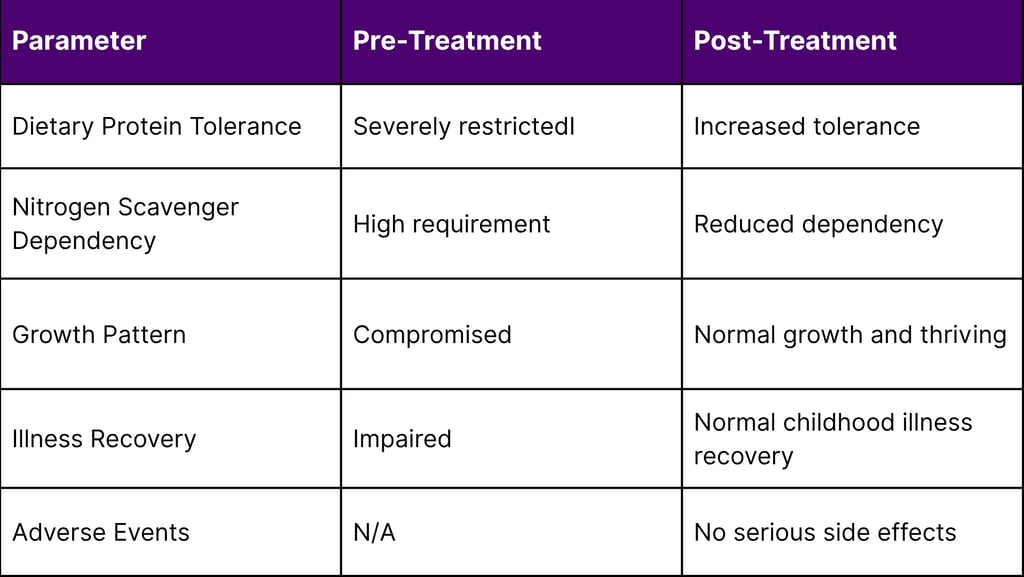
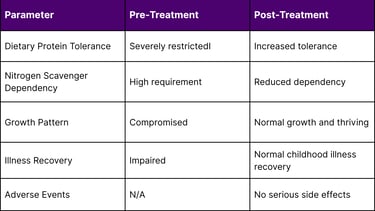
The treatment has demonstrated an excellent safety profile throughout the treatment period. As of April 2025, KJ has received three doses of the therapy with no serious side effects. This safety record is particularly significant given that this was the first human application of personalized CRISPR therapy.
Clinical Improvements
The therapeutic outcomes have exceeded expectations across multiple clinical parameters:
Metabolic Function Enhancement
In the short time since treatment, he has tolerated increased dietary protein and needed less nitrogen scavenger medication. KJ demonstrated progressive improvements, including enhanced tolerance to increased dietary protein intake and decreased reliance on nitrogen scavenger medications.
Overall Health Status
The treatment was administered safely, and he is now growing well and thriving. Additionally, he has been able to recover from certain typical childhood illnesses, indicating improved overall metabolic stability.
Clinical Success Metrics
Implications for Personalized Medicine
Paradigm Shift in Rare Disease Treatment
This breakthrough represents a fundamental shift from traditional one-size-fits-all treatments to truly personalized therapeutic approaches. The ability to develop a customized treatment within six months from diagnosis to administration sets a new standard for rapid therapeutic development in rare diseases.
Technological Advancement
The success of this treatment validates several key technological advances:
Precision Base Editing: Demonstrated ability to make targeted genetic corrections in vivo
Rapid Development Pipeline: Six-month timeline from diagnosis to treatment
Liver-Targeted Delivery: Successful delivery of gene-editing components to hepatocytes
Safety Profile: No serious adverse events in first human application
Market Impact and Future Prospects
Addressing Unmet Medical Needs
CPS1 deficiency represents one of many rare genetic disorders with limited treatment options. CPS1 deficiency presents in the neonatal period with hyperammonemia, resulting in death or neurological sequelae if patients survive. The success of this personalized approach opens new possibilities for treating similar ultra-rare conditions.
Economic Considerations
While specific cost data for this breakthrough treatment is not yet available, the potential economic impact includes:
Reduced lifetime healthcare costs for affected families
Decreased burden on healthcare systems
Potential for expanded applications to other genetic disorders
Investment opportunities in personalized gene editing platforms
Regulatory and Development Landscape
Clinical Trial Framework
This treatment was conducted as an n-of-1 clinical trial for a CRISPR gene-editing strategy, representing a novel regulatory approach for ultra-rare diseases where traditional clinical trial designs may not be feasible.
Future Development Pathway
The success of this initial treatment paves the way for:
Expanded patient populations
Refined delivery mechanisms
Broader application to other urea cycle disorders
Enhanced manufacturing capabilities for personalized therapies
Scientific Leadership and Collaboration
The breakthrough was achieved through collaboration between leading academic institutions and research centers. Physician-scientists at CHOP discussed the CRISPR gene-editing strategy, highlighting the importance of multidisciplinary expertise in advancing personalized medicine.
Conclusion: A New Era in Genetic Medicine
The successful treatment of CPS1 deficiency using personalized CRISPR gene editing represents more than just a medical breakthrough—it signifies the dawn of a new era in genetic medicine. This achievement demonstrates that even the rarest genetic disorders can be addressed through innovative, personalized approaches when cutting-edge science meets clinical expertise.
As we move forward, this pioneering treatment serves as a blueprint for addressing other rare genetic conditions, offering hope to families facing similar diagnoses and establishing new standards for rapid, personalized therapeutic development. The implications extend far beyond CPS1 deficiency, potentially transforming how we approach the entire spectrum of genetic diseases.
The success of this treatment validates the investment in personalized medicine technologies and sets the stage for continued innovation in gene editing therapies. As the field advances, we can anticipate more rapid development timelines, enhanced safety profiles, and broader applications that will benefit patients worldwide.
FAQs
Q1: What exactly is CPS1 deficiency and how common is it?
A: CPS1 deficiency is a rare genetic disorder affecting the urea cycle, the body's process for removing toxic ammonia from the blood. It occurs in approximately 1 in 526,000 to 1.3 million live births globally, making it an ultra-rare condition. The disorder is caused by mutations in the CPS1 gene, which provides instructions for making the enzyme carbamoyl phosphate synthetase 1, essential for ammonia detoxification in the liver.
Q2: How does this personalized CRISPR treatment differ from traditional gene therapies?
A: Unlike traditional gene therapies that use a standardized approach for all patients, this breakthrough treatment was completely customized for the individual patient's specific genetic mutations. The therapy was developed from scratch in just six months, using base editing technology to make precise corrections to the patient's DNA directly in liver cells. This represents the first successful application of fully personalized CRISPR therapy in humans.
Q3: What were the key safety considerations for this first-in-human personalized treatment?
A: Safety was paramount given this was the first personalized CRISPR application. The treatment underwent rigorous preclinical testing and was conducted as an n-of-1 clinical trial with extensive monitoring. As of April 2025, the patient has received three doses with no serious adverse events reported. The base editing approach was chosen specifically because it's less likely to cause unintended genetic changes compared to traditional CRISPR techniques.
Q4: Can this approach be applied to other rare genetic diseases?
A: Yes, this breakthrough has significant implications for treating other rare genetic disorders. The success demonstrates that personalized gene editing can be developed rapidly and safely, potentially benefiting patients with any of the estimated 7,000+ known rare diseases. The approach is particularly promising for conditions caused by single-gene mutations, especially those affecting the liver where gene editing delivery is more established.
Q5: What is the typical timeline and cost for developing such personalized treatments?
A: This case demonstrated remarkable speed, with development completed in just six months from diagnosis to first treatment. While specific costs haven't been disclosed, the approach represents a new model for rapid therapeutic development in ultra-rare diseases. The timeline and cost-effectiveness will likely improve as the technology matures and manufacturing processes become more streamlined.
Q6: How is the patient doing now, and what does long-term monitoring involve?
A: As of the latest reports in April 2025, the patient (KJ) is thriving with normal growth and development. He has demonstrated improved tolerance to dietary protein, reduced dependence on nitrogen scavenger medications, and enhanced ability to recover from typical childhood illnesses. Long-term monitoring will continue to assess both the durability of the treatment effects and any potential long-term safety considerations.
Q7: What regulatory pathway was used for this treatment, and how might this influence future approvals?
A: The treatment was conducted under an n-of-1 clinical trial framework, which is designed for situations where traditional clinical trial designs aren't feasible due to the ultra-rare nature of the condition. This approach could serve as a model for future personalized therapies, potentially accelerating access to life-saving treatments for patients with rare genetic disorders.
Q8: What role did artificial intelligence and computational tools play in developing this therapy?
A: While specific details about computational approaches haven't been fully disclosed, modern personalized gene editing relies heavily on sophisticated bioinformatics tools for designing precise genetic corrections, predicting off-target effects, and optimizing delivery systems. The rapid six-month development timeline likely benefited from advanced computational modeling and AI-assisted design processes.
References
U.S. Food and Drug Administration (FDA) – Orphan Drug Designations and Approvals.
European Medicines Agency (EMA) – Orphan Medicines.
National Institutes of Health (NIH) – Rare Diseases Research & Support (NCATS).
World Health Organization (WHO) – Global Health Estimates and Rare Disease Impact. T
Children's Hospital of Philadelphia Research Institute – Peer-reviewed clinical documentation of CPS1 deficiency treatment outcomes, February-April 2025.
Nature Medicine – Base Editing Technologies and Clinical Applications. Published clinical studies and systematic reviews, 2024-2025.
New England Journal of Medicine – Peer-reviewed analysis of n-of-1 trial methodologies, 2024.
American Journal of Human Genetics – Comprehensive review of urea cycle disorders and emerging treatments, 2024.
Cell – Technical review of base editing technologies and safety profiles in clinical applications, 2024.
Orphanet Journal of Rare Diseases – Meta-analysis of prevalence, mortality, and clinical outcomes data, 2024.
This breakthrough treatment was developed and administered at Children's Hospital of Philadelphia, representing a collaborative effort between academic researchers, clinicians, and biotechnology innovators. The patient continues to be monitored as part of ongoing clinical evaluation.
Navigation
© 2026 FyreIgnis Market Research. All rights reserved.
Legal
info@fyreignismarketresearch.com
India
