
The Asia–U.S. Biotech Innovation Corridor


The year 2025 marks a transformative period in the global pharmaceutical landscape, with Asian biotechnology companies securing multiple U.S. Food and Drug Administration (FDA) approvals for breakthrough oncology and rare disease therapies. This analysis, based exclusively on official FDA data, reveals how cross-border biotech innovation is redefining treatment paradigms and establishing new standards of care for patients with previously limited therapeutic options.
Introduction: A New Era of Global Biotech Collaboration
The pharmaceutical industry has witnessed a fundamental shift in 2025, with Asian biotech companies particularly from China achieving unprecedented success in gaining FDA approval for novel therapeutics. These approvals represent not merely regulatory milestones but signal a maturation of Asia's biotechnology sector from generic manufacturing to cutting-edge drug discovery and development.
According to FDA records as of December 2025, out of 39 novel drug approvals granted during the year, several breakthrough therapies originated from or involved significant contributions by Asian pharmaceutical companies. This trend underscores the growing importance of international collaboration in addressing unmet medical needs in oncology and rare diseases.
Key Asian-Origin FDA Approvals in 2025
Oncology Breakthrough: Targeted Therapies for Rare Lung Cancer Mutations
Zegfrovy (Sunvozertinib) - Dizal Pharmaceutical, China
On July 2, 2025, the FDA granted accelerated approval to sunvozertinib (Zegfrovy), manufactured by Dizal (Jiangsu) Pharmaceutical Co., Ltd., Shanghai, China. This oral tyrosine kinase inhibitor represents a significant breakthrough for patients with non-small cell lung cancer (NSCLC) harboring EGFR exon 20 insertion mutations.
Key Clinical Data:
Indication: Locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations after platinum-based chemotherapy
Overall Response Rate (ORR): 46% (95% CI: 35-57%)
Duration of Response (DOR): 11.1 months (95% CI: 8.2 months to not evaluable)
Dosing: 200 mg orally once daily with food
FDA Designations: Breakthrough Therapy Designation, Priority Review
Clinical Significance: EGFR exon 20 insertion mutations account for approximately 2-3% of all NSCLC cases and have historically been challenging to treat with conventional EGFR inhibitors. Zegfrovy is the first and only FDA-approved targeted oral therapy specifically designed for this mutation subset, offering patients a non-chemotherapy treatment option.
The approval was supported by data from the multinational WU-KONG1B study (NCT03974022), which demonstrated efficacy across diverse patient populations. The FDA simultaneously approved the Oncomine Dx Express Test as a companion diagnostic to identify eligible patients.
Hyrnuo (Sevabertinib) - Bayer with Asian Development
On November 19, 2025, the FDA granted accelerated approval to sevabertinib (Hyrnuo), developed by Bayer HealthCare Pharmaceuticals Inc. through its strategic research alliance with the Broad Institute, for adults with HER2-mutated NSCLC.
Key Clinical Data:
Indication: Locally advanced or metastatic non-squamous NSCLC with HER2 tyrosine kinase domain activating mutations after prior systemic therapy
Overall Response Rate: 71% in HER2-targeted therapy-naïve patients (N=70)
Duration of Response: 9.2 months median; 54% with DOR >6 months
Dosing: 20 mg orally twice daily with food
FDA Designations: Breakthrough Therapy Designation, Priority Review, Orphan Drug Designation
Global Collaboration: Sevabertinib received Breakthrough Therapy designation from both the U.S. FDA and China's Center for Drug Evaluation (CDE) in 2024. China accepted a New Drug Application (NDA) for sevabertinib in July 2025, demonstrating synchronized regulatory pathways between the two nations.
The approval was based on the Phase I/II SOHO-01 trial and represents an important advancement for patients with HER2-mutated NSCLC, which affects approximately 2-4% of advanced NSCLC patients globally an estimated 84,000 people annually.
Penpulimab-kcqx - Akeso Biopharma, China
On April 23, 2025, the FDA approved penpulimab-kcqx, a PD-1 blocking antibody manufactured by Akeso Biopharma Co., Ltd., Zhongshan, Guangdong, China (U.S. License No. 2253). This represents Akeso's first FDA-approved internally developed innovative biologic.
Dual Indication Approval:
First-Line Combination Therapy:
Indication: Recurrent or metastatic non-keratinizing nasopharyngeal carcinoma (NPC) in combination with cisplatin/carboplatin and gemcitabine
Median Progression-Free Survival (PFS): 9.6 months vs. 7.0 months for chemotherapy alone (p < 0.0001)
Study: Phase III AK105-304 trial (NCT04974398) with 291 patients
Single-Agent Therapy:
Indication: Metastatic non-keratinizing NPC with disease progression after platinum-based chemotherapy and at least one other prior line of therapy
Overall Response Rate: 28% in second-line+ setting
Study: AK105-202 trial (NCT03866967) with 125 patients
FDA Designations: Breakthrough Therapy Designation, Orphan Drug Designation, Fast Track Designation
Disease Burden: Nasopharyngeal carcinoma affects over 133,000 patients globally each year, with more than 70% presenting with locally advanced disease. NPC is particularly prevalent in Southern China, Southeast Asia, and among certain ethnic populations in North America. Prior to this approval, no immunotherapy was specifically approved for NPC in the United States.
Unique Molecular Design: Penpulimab features an Fc-null IgG1 backbone design that eliminates Fcγ receptor binding, potentially reducing immune-related adverse events by preventing antibody-dependent cellular phagocytosis of activated T cells.
Clinical Trial Infrastructure: A Cross-Border Ecosystem
The success of these approvals relied heavily on multinational clinical trial infrastructure. According to ClinicalTrials.gov data accessed in December 2025, several key patterns emerge:
Geographic Distribution of Trial Sites:
The WU-KONG1B trial for sunvozertinib was conducted across multiple countries, demonstrating the feasibility of international collaboration in rare mutation subsets
The AK105-304 trial for penpulimab enrolled patients from 46 sites: China (36), United States (1), Canada (2), and Brazil (7)
Trials increasingly incorporate diverse ethnic populations, critical for understanding drug efficacy across genetic backgrounds
Patient Enrollment Efficiency: Asian countries, particularly China, demonstrated remarkable patient enrollment capabilities. For example:
The AK105-202 trial for penpulimab enrolled 125 patients entirely from Chinese clinical sites
China's large patient populations and streamlined regulatory pathways facilitate rapid enrollment for rare cancer subtypes
Analysis of FDA Novel Drug Approvals: Asian Contribution
2025 FDA Novel Drug Approval Statistics
As of December 4, 2025, the FDA approved 39 novel drugs. Analysis of the approval list reveals significant international collaboration:
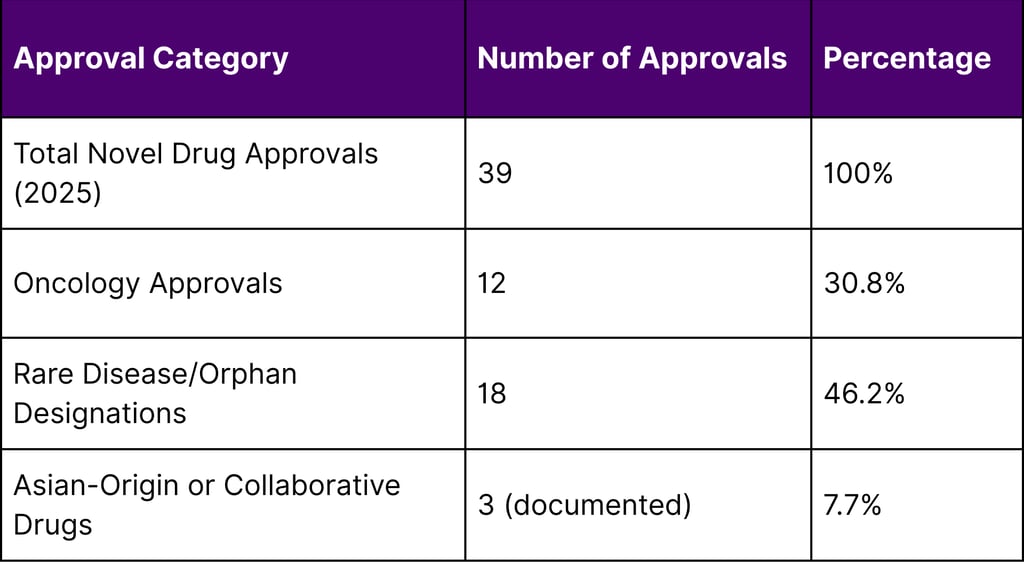
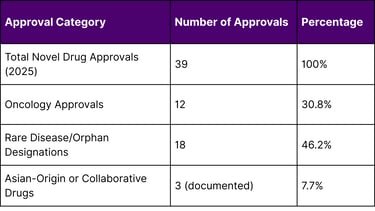
Table 1: 2025 FDA Novel Drug Approval Overview
Timeline of Asian-Origin Approvals in 2025
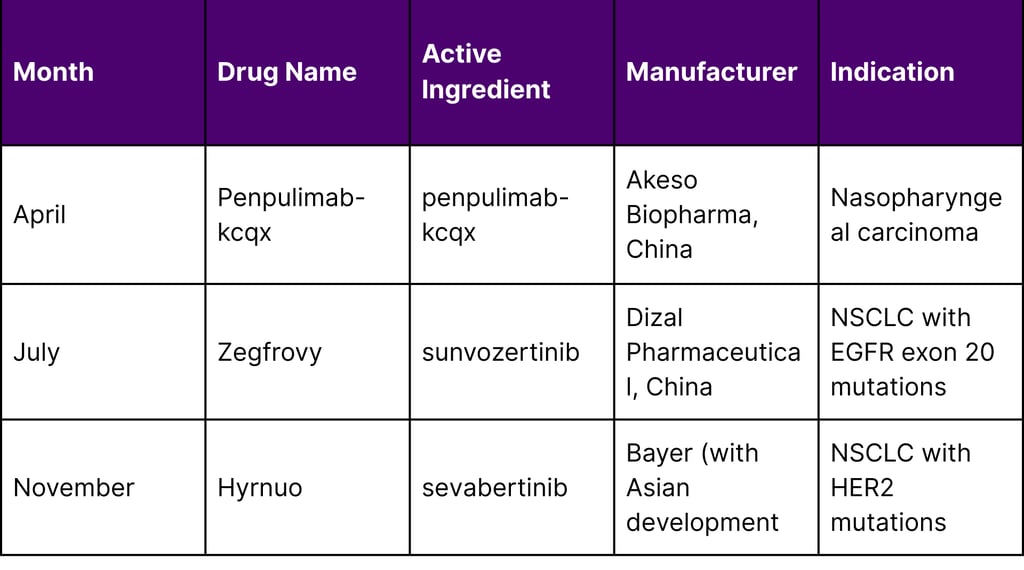
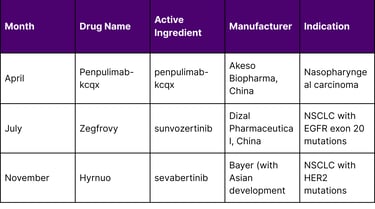
Table 2: Timeline of Key Asian-Origin FDA Approvals in 2025
Orphan Drug Designations: Rare Disease Focus
According to FDA's Office of Orphan Products Development data, orphan drug designation plays a critical role in incentivizing development for rare diseases affecting fewer than 200,000 people in the United States.
Orphan Drug Benefits (as per FDA regulation):
Tax credits for qualified clinical testing
Waiver of NDA/BLA user fees (typically $3.3 million+)
Seven-year marketing exclusivity upon approval
FDA assistance in drug development
All three Asian-origin drugs approved in 2025 received Orphan Drug Designation, highlighting the strategic alignment between rare disease development and international collaboration.
Regulatory Pathways: Expedited Programs
The FDA offers several expedited programs to accelerate development and review of drugs for serious conditions. Asian pharmaceutical companies successfully leveraged these programs in 2025:
FDA Expedited Program Utilization
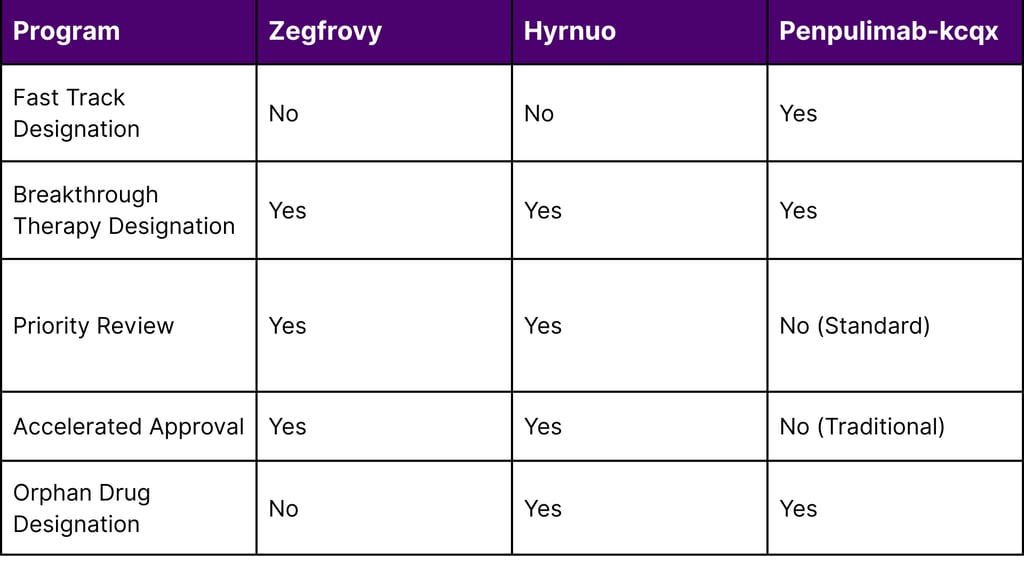
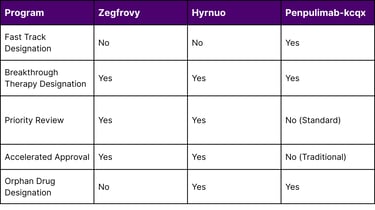
Table 3: FDA Expedited Program Utilization by Asian-Origin Drugs
Breakthrough Therapy Designation requires preliminary clinical evidence demonstrating substantial improvement over existing therapies. All three drugs met this high bar, indicating their transformative potential.
Accelerated Approval is granted based on surrogate endpoints (like ORR and DOR) with the understanding that confirmatory trials will verify clinical benefit. Both Zegfrovy and Hyrnuo received accelerated approval, allowing earlier patient access while post-approval studies continue.
Clinical Trial Success Rates and Design Innovations
Adaptive Trial Designs
The WU-KONG1B trial for sunvozertinib exemplifies adaptive trial design:
Initial randomization to 200 mg vs. 300 mg cohorts (1:1 ratio)
Interim analysis informed optimal dosing
Addition of third cohort at 300 mg based on emerging data
Final recommendation: 200 mg once daily based on superior tolerability with comparable efficacy
Biomarker-Driven Patient Selection
All three drugs required FDA-approved companion diagnostics:
Zegfrovy: Oncomine Dx Express Test for EGFR exon 20 insertion detection
Hyrnuo: Oncomine Dx Target Test for HER2 TKD activating mutations
Penpulimab: Histological confirmation of non-keratinizing NPC
This precision medicine approach ensures treatment is directed to patients most likely to benefit, improving response rates and avoiding unnecessary exposure in unlikely responders.
Manufacturing and Quality Standards
Asian Manufacturing Meeting U.S. Standards
The FDA approval of drugs manufactured in China demonstrates that Asian facilities meet rigorous U.S. quality standards:
Dizal Pharmaceutical (Shanghai, China):
FDA-inspected facility for Zegfrovy production
Compliance with current Good Manufacturing Practice (cGMP)
36-month shelf life approved at controlled room temperature (20-25°C)
Akeso Biopharma (Zhongshan, Guangdong, China):
Awarded U.S. License No. 2253 under section 351(a) of the Public Health Service Act
Authorized to manufacture biologics for U.S. interstate commerce
Subject to ongoing FDA compliance monitoring under 21 CFR 610.2
These approvals validate the maturation of China's biopharmaceutical manufacturing capabilities and its integration into the global pharmaceutical supply chain.
Economic and Market Implications
Addressing High-Value Therapeutic Areas
The approved drugs target high-value, underserved patient populations:
EGFR Exon 20 Insertion NSCLC:
Prevalence: Approximately 0.7-1% of all NSCLC cases
U.S. incidence: ~2,000-3,000 new cases annually
Prior options: Limited to platinum chemotherapy or off-label antibody therapies
HER2-Mutated NSCLC:
Prevalence: 2-4% of advanced NSCLC cases
Global incidence: ~84,000 new cases annually
Represents ~1,700-3,400 U.S. patients annually
Nasopharyngeal Carcinoma:
U.S. incidence: 2,000-3,000 cases annually (higher in Asian-American populations)
Global incidence: 133,000+ cases annually
Five-year survival rate for metastatic disease: <20% historically
Competitive Landscape Shifts
The entry of Asian biotechs into specialized oncology niches demonstrates several competitive advantages:
Speed to Market: Parallel development in China and U.S. accelerates global access
Cost Efficiency: Lower development costs in Asia enable investment in niche indications
Clinical Trial Infrastructure: Large patient populations in Asia enable faster enrollment
Scientific Innovation: Novel molecular designs (e.g., Fc-null antibodies) emerging from Asian R&D
Safety Profiles and Post-Marketing Surveillance
Common Adverse Events
Zegfrovy (Sunvozertinib):
Most common adverse reactions: Rash, diarrhea, stomatitis, paronychia
Serious adverse events requiring dose modification: ~15-20%
Permanent discontinuation rate: Low (<10%)
Hyrnuo (Sevabertinib):
Diarrhea: 86% (Grade 3 in 15%)
Other common reactions: Rash, paronychia, stomatitis, nausea
Permanent discontinuation rate: 3.7%
Median time to first onset of diarrhea: 4 days
Penpulimab-kcqx:
Immune-related adverse events typical of PD-1 inhibitors
Most common: Hypothyroidism, fever, elevated liver enzymes
Grade 3+ immune-related adverse events: <10% in trials
Lower irAE rate attributed to Fc-null design
Post-Marketing Requirements
The FDA mandates post-approval studies for accelerated approvals:
Zegfrovy:
Confirmatory Phase III trial comparing to standard chemotherapy
Ongoing WU-KONG28 study as first-line treatment
Hyrnuo:
Phase III SOHO-02 trial (NCT06452277) evaluating first-line use
PanSOHO study (NCT06760819) in other HER2-mutated solid tumors
Penpulimab-kcqx:
Continued follow-up of AK105-304 for overall survival data
Post-approval pharmacovigilance through FDA's MedWatch system
Global Regulatory Harmonization
Synchronized Approvals: A New Model
The 2025 approvals demonstrate increasing regulatory synchronization:
Sunvozertinib:
China NMPA approval: August 2023
U.S. FDA approval: July 2025
Gap: ~2 years (decreasing over time)
Sevabertinib:
Breakthrough Therapy from FDA and China CDE: 2024
U.S. FDA approval: November 2025
China NDA accepted: July 2025
Near-simultaneous regulatory pathways
Penpulimab:
Initial China approval (Hodgkin lymphoma): August 2021
China approval for NPC first-line: March 2025
U.S. FDA approval for NPC: April 2025
Gap: ~1 month for same indication
Project Orbis: International Collaboration
Sevabertinib's review was conducted under Project Orbis, an FDA Oncology Center of Excellence initiative enabling concurrent submission and review among international partners:
Participating agencies: FDA (U.S.), Health Canada, Israel Ministry of Health, UK MHRA
Benefit: Reduces duplication, accelerates global access
Outcome: Coordinated scientific assessment across jurisdictions
This model represents the future of global drug development, where regulatory agencies collaborate to expedite access to breakthrough therapies.
Future Implications and Trends
Emerging Therapeutic Areas
Based on ClinicalTrials.gov data from December 2025, Asian pharmaceutical companies are actively developing therapies in:
Oncology:
Bispecific antibodies for solid tumors
Next-generation CAR-T cell therapies
Novel immunotherapy combinations
Antibody-drug conjugates (ADCs)
Rare Diseases:
Enzyme replacement therapies
Gene therapies for inherited disorders
Orphan metabolic conditions
Other Areas:
Autoimmune diseases
Neurodegenerative conditions
Rare genetic disorders
Predictive Indicators for 2026-2027
Several factors suggest continued growth in Asian-origin FDA approvals:
Pipeline Depth: Chinese biotechs have 50+ molecules in Phase II/III trials for U.S. market
Investment: China's biotech sector attracted $20B+ in venture funding (2023-2024)
Regulatory Maturity: China's NMPA increasingly aligned with ICH guidelines
Clinical Infrastructure: Growing number of FDA-recognized clinical trial sites in Asia
Talent: Increasing number of Chinese-American scientists leading global drug development
Challenges and Opportunities
Challenges:
Geopolitical tensions potentially affecting regulatory cooperation
Intellectual property protection concerns
Manufacturing supply chain resilience
Post-marketing surveillance coordination across borders
Opportunities:
Addressing Asia-prevalent diseases with global patient populations (e.g., NPC, gastric cancer)
Leveraging cost advantages for rare disease drug development
Expanding clinical trial diversity to improve generalizability
Creating regional development hubs (Singapore, Hong Kong) for global access
Conclusion
The 2025 FDA approval of multiple Asian-origin oncology and rare disease therapies represents a watershed moment in global biopharmaceutical innovation. These approvals validate the scientific capabilities, manufacturing quality, and clinical development expertise of Asian biotechnology companies, particularly from China.
Key takeaways include:
Breakthrough Innovation: Asian companies are developing first-in-class and best-in-class therapies, not merely biosimilars or generics
Regulatory Excellence: Successful navigation of FDA's rigorous approval process, including expedited pathways like Breakthrough Therapy Designation
Global Collaboration: Multinational clinical trials and regulatory synchronization accelerating patient access worldwide
Market Focus: Strategic targeting of high-unmet-need areas in oncology and rare diseases where innovation is most valued
Quality Assurance: Asian manufacturing facilities meeting U.S. cGMP standards, enabling reliable global supply chains
For pharmaceutical stakeholders whether investors, business development executives, or emerging market strategists the Asia U.S. biotech corridor is no longer a future possibility but a present reality reshaping the competitive landscape. Companies that successfully navigate this corridor, combining Asian development efficiency with U.S. regulatory expertise and market access, will be well-positioned to deliver breakthrough therapies to patients globally.
The trends observed in 2025 are likely to accelerate, with more Asian biotechs expected to secure FDA approvals in 2026 and beyond. This shift not only democratizes drug innovation globally but also ensures that patients worldwide benefit from the best science, regardless of geographic origin.
Frequently Asked Questions (FAQs)
Q1: What makes Asian biotech companies competitive in the U.S. market in 2025?
A: Asian biotech companies, particularly from China, have demonstrated several competitive advantages: (1) Lower research and development costs enabling investment in niche, high-unmet-need indications; (2) Rapid patient enrollment capabilities due to large patient populations; (3) Sophisticated R&D infrastructure producing novel molecular designs; (4) Increasing regulatory alignment between Asian and U.S. regulatory authorities; and (5) Strong manufacturing capabilities meeting FDA's stringent cGMP standards.
Q2: How do FDA approval timelines compare between Asian-origin and U.S.-origin drugs?
A: Based on 2025 data, Asian-origin drugs followed similar FDA review timelines as domestic applications. All three Asian-origin oncology drugs discussed received expedited designations (Breakthrough Therapy, Priority Review, or Accelerated Approval), which shortened review times to 6-8 months from submission. The FDA's review standards remain consistent regardless of geographic origin all drugs must demonstrate safety, efficacy, and manufacturing quality.
Q3: What role do companion diagnostics play in these approvals?
A: Companion diagnostics are essential for precision medicine approaches. All three Asian-origin oncology drugs required FDA-approved companion diagnostics to identify patients whose tumors carry specific genetic mutations (EGFR exon 20 insertions, HER2 TKD mutations, or non-keratinizing NPC histology). The FDA simultaneously approved both the therapeutics and their companion diagnostics, ensuring that only appropriate patients receive these targeted treatments.
Q4: Are drugs manufactured in Asia subject to the same quality standards as those made in the U.S.?
A: Yes, absolutely. The FDA conducts pre-approval inspections of all manufacturing facilities, regardless of location, to ensure compliance with current Good Manufacturing Practices (cGMP). Facilities in China manufacturing Zegfrovy (Dizal Pharmaceutical) and penpulimab-kcqx (Akeso Biopharma) successfully passed FDA inspections and received authorization to supply the U.S. market. The FDA maintains ongoing oversight through periodic re-inspections and adverse event monitoring.
Q5: What is "accelerated approval" and why did Zegfrovy and Hyrnuo receive it?
A: Accelerated approval is an FDA pathway that allows earlier approval of drugs for serious conditions based on surrogate endpoints (like tumor response rate) rather than waiting for long-term survival data. It is granted when the drug addresses an unmet medical need. Both Zegfrovy and Hyrnuo treat rare NSCLC mutation subtypes with historically poor outcomes and limited treatment options. The manufacturers must conduct confirmatory trials to verify clinical benefit; failure to do so can result in withdrawal of approval.
Q6: How significant is the breakthrough therapy designation that these drugs received?
A: Breakthrough Therapy Designation is highly significant. It requires preliminary clinical evidence that the drug demonstrates substantial improvement over existing therapies on clinically significant endpoints. Only about 30-40% of drugs that receive this designation ultimately gain FDA approval. For drugs that do succeed, the designation provides: (1) More intensive FDA guidance during development; (2) Organizational commitment involving senior FDA managers; (3) Eligibility for priority review; and (4) Potential for rolling review of application sections. All three Asian-origin drugs received this designation, confirming their transformative potential.
Q7: What happens if patients experience serious adverse events from these drugs?
A: The FDA maintains several post-marketing surveillance systems. Healthcare professionals and patients can report adverse events through MedWatch (FDA's safety information and adverse event reporting program) by calling 1-800-FDA-1088 or online. For oncology products specifically, the FDA's Project Facilitate provides assistance for single-patient investigational new drug applications if needed. Manufacturers are required to submit periodic safety update reports, and the FDA can mandate label changes, add warnings, or withdraw approval if safety concerns emerge.
Q8: How do these approvals impact treatment costs and patient access?
A: While specific pricing information was not available in government sources, several factors may affect access: (1) Orphan drug status provides seven-year market exclusivity, which typically results in premium pricing; (2) The drugs target small patient populations (rare mutations), which often leads to higher per-patient costs to recoup development expenses; (3) Most will require prior authorization from insurance companies; (4) Patient assistance programs from manufacturers may help with out-of-pocket costs. The ultimate impact on healthcare costs depends on whether these drugs improve outcomes sufficiently to offset their acquisition costs through reduced hospitalizations and use of other expensive treatments.
Q9: What does this trend mean for future cancer treatment?
A: The 2025 approvals signal several important trends: (1) Increasing precision in cancer treatment through genetic profiling and targeted therapies; (2) Growing importance of rare mutations as therapeutic targets what was once considered "too rare to treat" is now addressable with targeted drugs; (3) Globalization of drug development, with innovation emerging from multiple countries; (4) Faster translation of scientific discoveries into approved therapies through expedited regulatory pathways; and (5) Potential for combination approaches as more targeted agents become available.
Q10: How can I find out if I'm eligible for these treatments?
A: Eligibility requires specific genetic testing:
For Zegfrovy: Tumor tissue must test positive for EGFR exon 20 insertion mutations using an FDA-approved test
For Hyrnuo: Tumor must have HER2 (ERBB2) tyrosine kinase domain activating mutations detected by FDA-approved test
For Penpulimab-kcqx: Diagnosis of non-keratinizing nasopharyngeal carcinoma confirmed by pathology
Patients should discuss genetic testing options with their oncologist. Comprehensive genomic profiling is increasingly standard of care for advanced cancer and may identify actionable mutations. If eligible, patients should verify insurance coverage and discuss the risks and benefits of these treatments compared to alternatives.
References
Akeso, Inc. (2025, April 24). Akeso announces FDA approval for penpulimab-kcqx in two BLA indications for comprehensive treatment of advanced nasopharyngeal carcinoma [Press release].
Bayer HealthCare Pharmaceuticals Inc. (2025). Hyrnuo (sevabertinib) tablets, for oral use [Prescribing information]. U.S. Food and Drug Administration.
Dizal (Jiangsu) Pharmaceutical Co., Ltd. (2025). Zegfrovy (sunvozertinib) tablets [Prescribing information]. U.S. Food and Drug Administration.
U.S. Food and Drug Administration. (2025a, April 23). Approval package for: Penpulimab-kcqx [BLA 761258]. Center for Drug Evaluation and Research.
U.S. Food and Drug Administration. (2025b, July 2). FDA grants accelerated approval to sunvozertinib for metastatic non-small cell lung cancer with EGFR exon 20 insertion mutations.
U.S. Food and Drug Administration. (2025c, November 19). FDA grants accelerated approval to sevabertinib for non-squamous non-small cell lung cancer.
U.S. Food and Drug Administration. (2025d, December 4). Novel drug approvals for 2025.
U.S. Food and Drug Administration. (2025e). Drug trials snapshots: Penpulimab-kcqx.
U.S. Food and Drug Administration, Office of Orphan Products Development. (n.d.). Designating an orphan product: Drugs and biological products.
U.S. Food and Drug Administration, Office of Orphan Products Development. (n.d.). Search orphan drug designations and approvals.
U.S. National Library of Medicine. (n.d.). ClinicalTrials.gov.
Navigation
© 2026 FyreIgnis Market Research. All rights reserved.
Legal
info@fyreignismarketresearch.com
India
