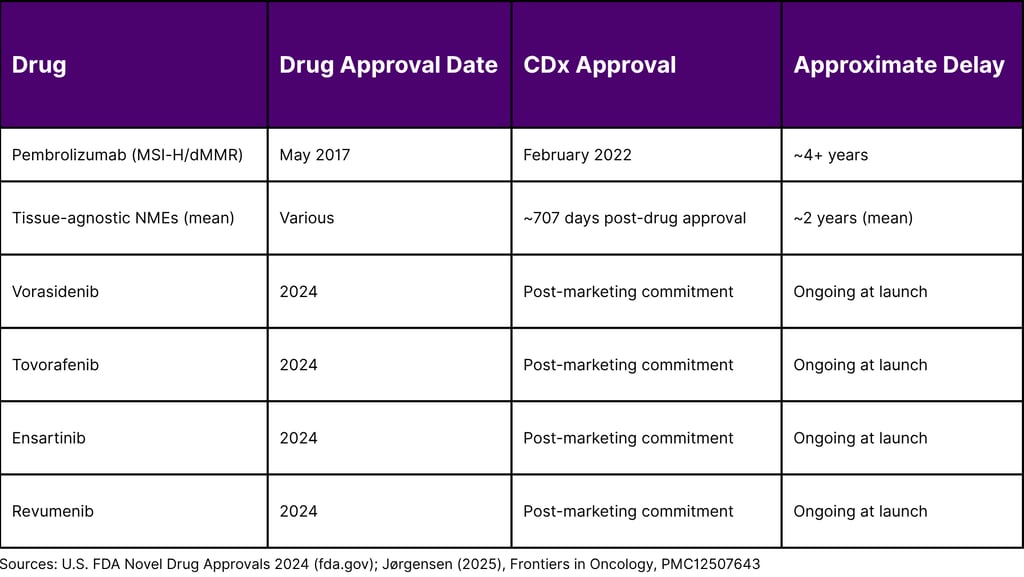
In 2024, four notable drugs vorasidenib, tovorafenib, ensartinib, and revumenib reached the market carrying label language explicitly stating that no FDA-approved CDx was currently available (FDA, 2024). All four subsequently had companion diagnostics approved post-launch, highlighting the gap between package insert language and the evolving CDx approval database.
Across 2024 and 2025 combined, approximately one in three novel oncology drug approvals reached the market without a simultaneously approved CDx (FDA CDx database; Jørgensen, 2025).
The Regulatory Framework: Flexibility by Design, Complexity in Practice
The FDA's 2014 final guidance, In Vitro Companion Diagnostic Devices, was designed to encourage early-stage collaboration between drug and diagnostic developers, with the stated goal of "faster access to promising new treatments for patients living with serious and life-threatening diseases" (FDA, 2014). This was supplemented by the 2016 draft guidance, Principles for Codevelopment of an In Vitro Companion Diagnostic Device with a Therapeutic Product, which laid out practical frameworks for parallel development.
Critically, FDA guidance does call for contemporaneous drug and diagnostic development but it has also accepted post-marketing commitments as a common and workable pathway. There is no public record of drug approval being withheld solely due to CDx delays (with one historical exception) (FDA Companion Diagnostics page, 2024).
This regulatory flexibility is, in principle, patient-friendly: it prevents CDx development from becoming a bottleneck to drug approval. In practice, however, it creates a post-approval commercialisation gap that is costly for pharmaceutical companies and disruptive for market access teams.
The classification question is also shifting. On 25 November 2025, the FDA published a proposed order in the Federal Register to reclassify certain oncology nucleic acid-based CDx tests from Class III medical devices (requiring full Premarket Approval, or PMA) to Class II devices (subject to the faster, lower-cost 510(k) pathway) (Federal Register, Vol. 90, 2025). If finalised, this reclassification would materially reduce the regulatory burden and timeline for CDx approval, potentially narrowing the launch gap that commercial teams currently navigate.
Commercial Consequences: The Cost of the Gap
The biomarker bottleneck is not merely a regulatory inconvenience it carries direct commercial consequences that market access and launch planning teams must quantify and mitigate.
Patient identification is constrained. Without an approved, widely deployed CDx, prescribers may rely on laboratory-developed tests (LDTs), which vary in analytical performance, reimbursement status, and clinical site availability. LDT use can result in inconsistent patient identification, uneven market penetration, and post-launch revenue shortfalls that are difficult to reverse.
Payer negotiations are complicated. CDx test costs are typically separate from the drug reimbursement pathway, requiring distinct health technology assessment (HTA) submissions and reimbursement approvals. Where a CDx lacks formal approval or consistent reimbursement, payer willingness to fund the associated drug therapy may be reduced or delayed.
Launch trajectory is affected. The standard oncology drug adoption ramp assumes that eligible patients can be reliably identified at launch. A CDx gap compresses the eligible population in the early launch window precisely the period when commercial investment is highest and when establishing prescriber habit is most critical.
The NCI's Molecular Analysis for Therapy Choice (NCI-MATCH) trial, the largest precision medicine trial of its kind, underscores just how central biomarker-directed patient matching has become to the clinical infrastructure: the trial specifically uses biomarker tests to assign patients to treatments based on genetic alterations in their tumours, across cancer types (NIH/NCI, 2025). The operational complexity of this at a trial scale maps directly onto the commercial challenges of doing the same at a market-wide level.
What This Means for Market Access Teams
For oncology market access professionals, the biomarker bottleneck demands a shift from reactive to proactive planning. Several strategic imperatives emerge:
1. CDx strategy must begin at Phase II. Waiting until Phase III or regulatory submission to formalise CDx co-development leaves commercial teams exposed. FDA guidance explicitly encourages early collaboration, and the data show that contemporaneous approval while not mandated is achievable when planning begins early.
2. Post-marketing commitment planning should be a launch input, not an afterthought. Where CDx approval is expected post-launch, market access teams should model the patient identification gap, plan for LDT bridging strategies, and set realistic uptake curves that reflect real-world diagnostic availability.
3. Reimbursement pathways for CDx must be mapped in parallel. Payer access for the drug and the diagnostic are linked but are frequently managed on different timelines. Misalignment creates avoidable market access delays.
4. The regulatory reclassification proposal warrants attention. If the FDA's November 2025 proposal to reclassify nucleic acid-based CDx tests from Class III to Class II is finalised, it could significantly accelerate CDx approval timelines and alter the commercial calculus for launches currently planned against a post-marketing CDx commitment.
5. Biomarker prevalence data should inform launch sequencing. For rare disease and biomarker-defined oncology populations, accurate epidemiological modelling grounded in genomic prevalence data is foundational to realistic revenue forecasting and market sizing.
The Path Forward: Narrowing the Gap
The FDA's Precision Oncology Program has stated its intent to "catalyse regulatory science research and development to apply new methodologies to deliver the promise of precision oncology" (FDA, Precision Oncology Program, 2025). The agency's June 2023 pilot programme for oncology drug products used with certain IVD tests represents a further step toward greater transparency around biomarker performance expectations.
Meanwhile, NCI continues to invest in the molecular diagnostic infrastructure required to support biomarker-driven patient matching at scale. NCI's Molecular Diagnostic Network (MDNet), launched to support precision medicine trials including MyeloMATCH, performs all molecular and cellular biomarker assays for NCI precision medicine programmes a model for the kind of integrated diagnostic infrastructure that commercial launches will increasingly need to replicate (NCI/DCTD, 2025).
The oncology sector is moving toward a future where CDx co-development is not just expected but operationally embedded from the earliest stages of drug development. The organisations both in pharma and in market research that build the analytical frameworks, the regulatory intelligence, and the commercial modelling to navigate CDx timing will be best positioned to protect launch value in an increasingly biomarker-driven landscape.
Conclusion
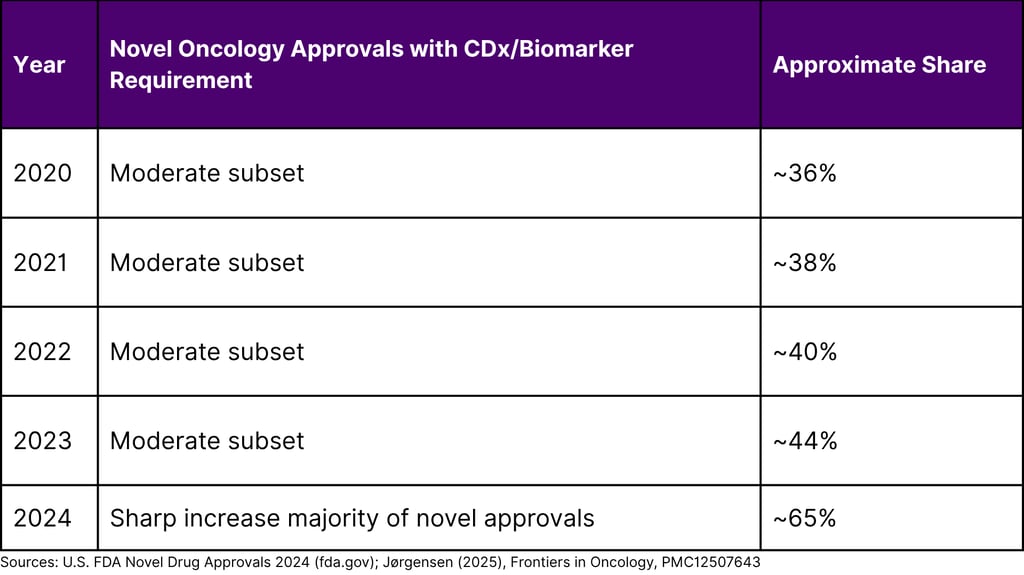
The biomarker bottleneck is one of oncology's most consequential and least publicly discussed commercial challenges. With 65% of novel oncology approvals in 2024 requiring a biomarker or CDx for patient selection, and roughly one in three of those drugs launching without a simultaneously approved diagnostic, the gap between science and commercial execution has never been more material.
For market access teams, the message is clear: CDx readiness is not a diagnostic company's problem alone. It is a shared commercial risk that must be built into launch strategy, payer engagement, revenue modelling, and regulatory planning from the earliest stages of drug development. The companies that recognise this and plan accordingly will be the ones whose launches fulfil their scientific promise.
FREQUENTLY ASKED QUESTIONS (FAQ)
Q1. What is a companion diagnostic (CDx)?
A companion diagnostic is a medical device typically an in vitro diagnostic that provides information essential for the safe and effective use of a corresponding drug or biological product. It is used to identify patients likely to benefit from a specific therapy, those at risk of serious side effects, or to monitor treatment response. The FDA maintains a regularly updated list of cleared or approved CDx devices on its website.
Q2. Why do companion diagnostics sometimes receive approval after the drug?
FDA guidance calls for contemporaneous co-development of drugs and their companion diagnostics, but the agency has accepted post-marketing commitments as a standard pathway when CDx development is not complete at the time of drug approval. This reflects deliberate regulatory flexibility to avoid CDx delays holding back patient access to potentially life-saving drugs.
Q3. What is the typical delay between drug approval and CDx approval?
Research published in Frontiers in Oncology (2025) found a mean delay of approximately 707 days (nearly two years) between drug approval and CDx approval for tissue-agnostic oncology NMEs approved up to 2025. Individual cases, such as pembrolizumab's MSI-H/dMMR CDx, have seen delays of more than four years.
Q4. What is the FDA's proposed reclassification of CDx tests, and why does it matter?
In November 2025, the FDA published a proposed order in the Federal Register to reclassify certain nucleic acid-based oncology CDx tests from Class III (requiring Premarket Approval) to Class II (subject to the 510(k) pathway). If finalised, this would reduce the regulatory burden, shorten review timelines, and likely encourage more diagnostic manufacturers to enter the CDx market which could meaningfully narrow the launch gap that commercial teams currently face.
Q5. What can market access teams do to reduce the impact of CDx launch delays?
Key actions include: initiating CDx co-development strategy at Phase II; incorporating post-marketing CDx commitment timelines into launch revenue models; developing LDT bridging strategies; pursuing parallel reimbursement pathways for both the drug and the CDx; and closely monitoring the FDA's evolving CDx regulatory framework, including the Class III-to-Class II reclassification proposal.
Q6. How prevalent is biomarker use in oncology clinical trials today?
According to the FDA's 2024 Drug Trials Snapshots Summary Report, three-quarters of all oncology clinical trials in 2024 included the use of a biomarker a figure that underscores how foundational biomarker-directed patient selection has become to modern oncology drug development.
REFERENCES
U.S. Food and Drug Administration. (2014). In vitro companion diagnostic devices: Guidance for industry and FDA staff. U.S. Department of Health and Human Services.
U.S. Food and Drug Administration. (2024). Novel drug approvals for 2024. U.S. Department of Health and Human Services.
U.S. Food and Drug Administration. (2024). List of cleared or approved companion diagnostic devices (in vitro and imaging tools). U.S. Department of Health and Human Services.
U.S. Food and Drug Administration. (2025). Precision Oncology Program. U.S. Department of Health and Human Services.
U.S. Food and Drug Administration. (2025). Companion diagnostics. U.S. Department of Health and Human Services.
U.S. Food and Drug Administration. (2025). Drug trials snapshots summary report 2024. U.S. Department of Health and Human Services.
U.S. Food and Drug Administration. (2025, November 25). Immunology and microbiology devices; reclassification of nucleic acid-based test systems for use with a corresponding approved oncology therapeutic product; proposed amendment; proposed order; request for comments. Federal Register, 90(226).
U.S. Food and Drug Administration. (2024). Specific test categories or technologies: Laboratory developed tests FAQs. U.S. Department of Health and Human Services.
Jørgensen, J. T. (2025). An analysis of FDA drug approvals for oncological hematological malignancies in relation to companion diagnostics. Frontiers in Oncology, 15, 1635491.
National Cancer Institute. (2025). Biomarker testing for cancer treatment. U.S. Department of Health and Human Services.
National Cancer Institute, Division of Cancer Treatment and Diagnosis. (2025). NCI precision medicine trials. U.S. Department of Health and Human Services.
National Institutes of Health. (2025). NIH clinical trial will test precision medicine treatments for myeloid cancers [Press release].
National Institutes of Health. (2018). NCI-MATCH precision medicine clinical trial releases new findings, strengthens path forward for targeted cancer therapies [Press release].