
The Rise of Ultra-Rare Disease Pipelines


The pharmaceutical landscape is witnessing a paradigm shift as ultra-rare disease treatments emerge as a lucrative frontier for innovation and investment. Once considered commercially unviable due to minuscule patient populations, these therapeutic areas are now attracting unprecedented attention from biopharmaceutical companies, investors, and regulatory bodies alike. With the FDA's continued commitment to accelerating approval pathways and supporting orphan drug development, the ultra-rare disease segment represents a goldmine of opportunity for those willing to navigate its unique challenges.
The Evolving Rare Disease Landscape
Understanding Rare and Ultra-Rare Diseases
According to the FDA, a rare disease is defined by the Orphan Drug Act as a disease that affects fewer than 200,000 people in the United States. Ultra-rare diseases typically affect even smaller populations—often fewer than 20,000 individuals, with some impacting only hundreds of patients globally.
The National Institutes of Health estimates that 25 to 30 million Americans are affected by rare diseases. Despite their individual rarity, these conditions collectively represent a substantial public health concern. There are thousands of rare diseases, with an estimated 85-90 percent considered serious or life-threatening. However, only a few hundred of these rare diseases currently have FDA approved treatments.
The Remarkable Progress in Orphan Drug Development
The landscape has transformed dramatically since the Orphan Drug Act was enacted in 1983. Before Congress enacted the 1983 law, there were only ten FDA approved treatments for orphan diseases. Since the law's passage, there have been more than 600 orphan drug indications approved.
This represents remarkable scientific and clinical progress for millions of patients affected by rare disorders. The momentum has only accelerated in recent years, as evidenced by the steady increase in orphan drug designation requests received by the Office of Orphan Products Development (OOPD). In 2016, OOPD received 568 new requests for designation – more than double the number of requests received in 2012.
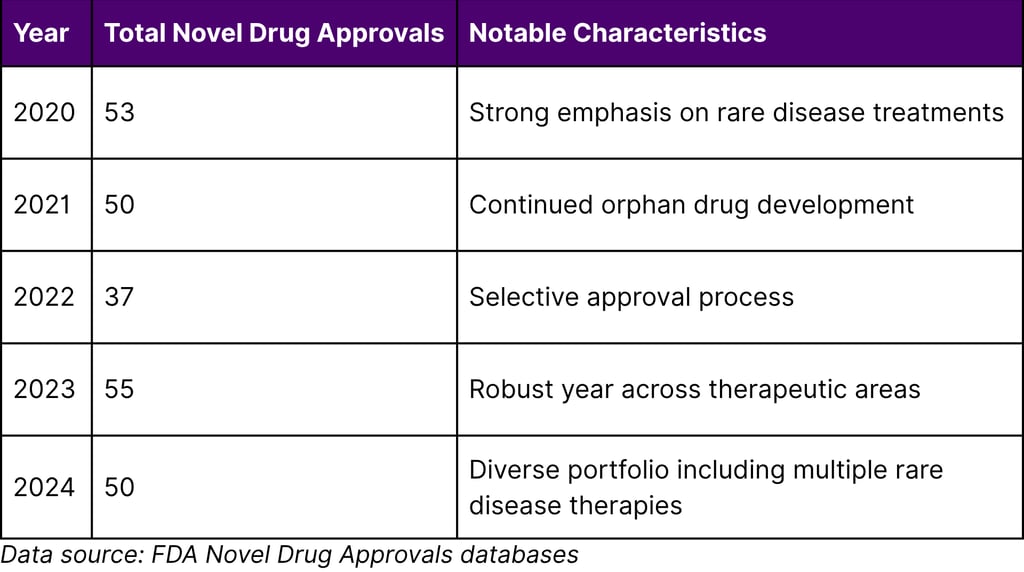
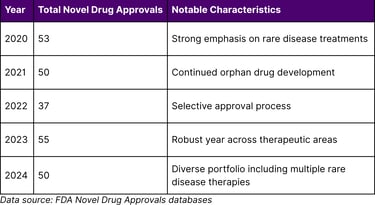
Recent FDA Approval Activity
The FDA has maintained robust drug approval activity in recent years:
2024 Novel Drug Approvals: The FDA approved 50 novel drugs in 2024, representing a diverse range of therapeutic areas and patient populations. Notable rare disease approvals included:
Treatments for Niemann-Pick disease type C (Aqneursa and Miplyffa)
Therapies for hemophilia A and B (Hympavzi and Alhemo)
Novel treatments for ultra-rare conditions like WHIM syndrome (Xolremdi)
Duchenne muscular dystrophy treatment (Duvyzat)
Multiple rare cancer therapies
FDA Approval Trends (2020-2024)
Regulatory Framework Revolution
The FDA has undertaken significant initiatives to streamline rare disease drug development:
September 2025 RDEP Framework: The FDA announced the Rare Disease Evidence Principles (RDEP), a transformative framework designed to accelerate approval pathways for treatments targeting ultra-rare genetic diseases. This initiative, jointly developed by the Center for Drug Evaluation and Research (CDER) and the Center for Biologics Evaluation and Research (CBER), provides unprecedented flexibility and predictability for developers working with extremely small patient populations.
FDA Rare Disease Innovation Hub: Launched in 2024, this hub serves as a single point of engagement that promotes cross-center dialogue on complex rare disease drug development issues. This structural innovation reflects the agency's commitment to reducing developmental barriers while maintaining rigorous safety standards.
Orphan Drug Modernization: In 2017, FDA launched its "90 in 90 Plan" to ensure timely review of orphan drug designation requests. The agency committed to respond to 100 percent of all new orphan drug designation requests within 90 days of their receipt, demonstrating its dedication to supporting rare disease drug development.
The Ultra-Rare Disease Definition and Strategic Importance
Defining Ultra-Rare Diseases
While the Orphan Drug Act establishes the threshold for rare diseases at fewer than 200,000 affected individuals in the United States, ultra-rare diseases represent an even smaller subset. These conditions typically affect:
Fewer than 20,000 individuals in the United States
In some cases, only hundreds or even dozens of patients globally
Predominantly genetic in origin, with identifiable molecular mechanisms
Often severe or life-threatening without available treatments
Key Characteristics of Ultra-Rare Disease Pipelines
Small Patient Populations: Recruiting sufficient patients for traditional clinical trials remains the primary challenge. The RDEP framework now accommodates single pivotal trials and innovative endpoint strategies to address this limitation.
Genetic Basis: Many ultra-rare diseases are driven by known genetic defects, enabling precision medicine approaches and biomarker-driven development strategies.
High Unmet Need: The absence of alternative treatments creates both ethical imperatives and commercial opportunities for breakthrough therapies.
Regulatory Advantages: The Orphan Drug Act provides for granting special status to a drug or biological product to treat a rare disease or condition, including benefits such as seven-year market exclusivity upon approval.
The Financial Appeal: Why Ultra-Rare Diseases Are Billion-Dollar Opportunities
Orphan Drug Designation Benefits
The FDA's orphan drug designation program provides substantial advantages to developers:
Market Exclusivity: Seven-year orphan exclusivity is granted upon marketing approval, determined by OOPD. This exclusivity is distinct from other exclusivities and can only be "broken" in cases of drug shortage or if another drug is clinically superior to the approved drug.
Development Incentives: The Orphan Drug Act provides multiple benefits including:
Tax credits for qualified clinical testing expenses
Waiver of FDA application fees
Potential for accelerated approval pathways
Access to FDA's orphan drug grant program
Long-Term Revenue Potential Through Label Expansion
While initial approval targets a rare disease indication, many orphan drugs achieve significant commercial success through strategic label expansion. Developers can pursue:
Multiple rare disease indications: Expanding within the rare disease space
Common disease applications: Leveraging the same mechanism of action for broader patient populations
Global market expansion: Securing approvals in multiple regions
The ability to command premium pricing for initial rare disease indications, combined with potential expansion opportunities, creates compelling revenue trajectories that can reach or exceed billion-dollar annual sales.
Government Investment and Support
Governments worldwide recognize the strategic importance of rare disease research. In the United States, the FDA continues to invest in:
The Rare Disease Endpoint Advancement (RDEA) Pilot Program
The Rare Disease Cures Accelerator Data and Analytics Platform (RDCA-DAP)
Natural history study support through OOPD grant programs
International collaboration through partnerships with the European Medicines Agency
Scientific and Technological Drivers
Genomic Medicine Revolution
Advances in next-generation sequencing and CRISPR gene-editing technologies have fundamentally transformed ultra-rare disease drug development. Genetic characterization enables:
Precision diagnosis: Reducing diagnostic odysseys from years to months
Biomarker identification: Facilitating smaller, more efficient clinical trials
Gene therapy approaches: Offering potential one-time curative treatments
Natural history studies: Providing control data without requiring traditional placebo arms
Innovative Clinical Trial Designs
The RDEP framework encourages novel approaches that would have been unthinkable a decade ago:
N-of-1 trials: Demonstrating efficacy in individual patients with unique genetic profiles
Basket trials: Grouping patients by molecular characteristics rather than anatomical disease classifications
Adaptive designs: Allowing protocol modifications based on emerging data
External control arms: Utilizing historical data and registry information to reduce patient burden
Drug Modality Diversity
The 2024 FDA approvals demonstrated viable pathways across multiple therapeutic modalities:
Gene therapies: One-time interventions with transformative potential
Enzyme replacement therapies: Long-established approach for metabolic disorders
Antisense oligonucleotides: Precision targeting of genetic messages
Small molecule therapies: Traditional oral medications for genetic conditions
Monoclonal antibodies: Targeting specific disease mechanisms
Key Therapeutic Areas in Ultra-Rare Disease Pipelines
Metabolic Disorders
Inborn errors of metabolism represent classic ultra-rare diseases with well-characterized genetic bases. Recent FDA approvals in this area include treatments for Niemann-Pick disease type C, demonstrating the viability of developing therapies for previously untreatable metabolic conditions.
Neurodegenerative and Neuromuscular Diseases
The approval of Duvyzat for Duchenne muscular dystrophy in 2024 exemplifies breakthrough progress in this therapeutic area. Neuromuscular diseases combine high unmet need with premium pricing potential and strong patient advocacy support.
Rare Hematologic Disorders
The FDA approved multiple hemophilia treatments in 2024, including Hympavzi and Alhemo, demonstrating continued innovation in rare blood disorders. These conditions benefit from decades of natural history data and established clinical networks.
Ultra-Rare Immunodeficiencies
The 2024 approval of Xolremdi for WHIM syndrome—an ultra-rare immunodeficiency affecting only a small number of patients globally—demonstrates that even the rarest conditions can attract successful drug development when the scientific rationale is strong.
Rare Cancers
The FDA continues to approve treatments for rare cancer types, leveraging accelerated approval pathways and orphan drug designation to bring novel therapies to patients with limited options.
Challenges and Risk Factors
Despite the enormous potential, ultra-rare disease drug development faces significant hurdles:
Clinical Development Barriers
Patient Recruitment: Identifying and enrolling patients globally remains resource-intensive. Developers must work with patient advocacy groups, establish international consortia, and leverage natural history registries.
Endpoint Selection: Natural history variability complicates outcome measure selection. The RDEP framework provides flexibility, but developers must still justify their chosen endpoints.
Trial Duration: Extended follow-up periods may be necessary to demonstrate durability, particularly for gene therapies and other novel modalities.
Manufacturing Complexity: Personalized therapies require novel production capabilities and supply chain management.
Regulatory Considerations
While the RDEP framework provides enhanced clarity, developers must navigate:
Evolving standards for real-world evidence integration
Post-marketing commitment requirements for accelerated approvals
Conversion requirements from accelerated to full approval
International regulatory harmonization challenges
Market Access and Reimbursement
Premium pricing strategies face increasing scrutiny:
Payer negotiations: High per-patient costs trigger intensive value assessments
Budget impact: Health systems must balance access with financial sustainability
Outcome-based agreements: Performance-linked payment models add complexity
Global access: International pricing strategies must accommodate diverse reimbursement environments
Scientific Risks
Mechanism validation: Novel targets may fail to translate from preclinical models to human patients.
Durability questions: Long-term efficacy data may take years to generate, particularly for gene therapies.
Safety signals: Small trial populations may miss important adverse events that only become apparent with broader use.
Biomarker limitations: Surrogate endpoints may not perfectly predict clinical benefit.
Strategic Considerations for Stakeholders
For Biopharmaceutical Companies
Early FDA Engagement: Leverage the RDEP framework and Rare Disease Innovation Hub for early regulatory guidance.
Partnership Strategies: Collaborate with patient advocacy groups to accelerate recruitment and enhance development insights.
Portfolio Diversification: Balance ultra-rare disease assets with broader indications to mitigate risk.
Manufacturing Planning: Invest in flexible, scalable production capabilities from early development stages.
For Investors
Due Diligence Focus: Evaluate scientific rationale, regulatory pathway clarity, and management execution capabilities.
Milestone-Based Investment: Structure capital deployment around regulatory and clinical inflection points.
Risk Assessment: Understand that ultra-rare disease programs carry both exceptional upside potential and meaningful development risks.
Exit Strategy Diversity: Recognize both acquisition potential and public market opportunities.
For Healthcare Systems
Early Assessment Frameworks: Develop expedited evaluation processes for breakthrough therapies.
Budget Planning: Anticipate orphan drug expenditures through pipeline monitoring and forecasting.
Multidisciplinary Evaluation: Engage clinical, ethical, and financial stakeholders in access decisions.
Outcome Tracking: Build infrastructure to monitor real-world effectiveness and safety.
Future Outlook: The Next Frontier
Technology Convergence
The integration of artificial intelligence, advanced genomics, and innovative manufacturing will further accelerate ultra-rare disease drug development:
AI-powered drug discovery: Identifying therapeutic candidates with unprecedented speed
Digital biomarkers: Enabling remote, continuous patient monitoring
Personalized manufacturing: Making individualized therapies economically viable
Predictive analytics: Optimizing patient selection and dosing strategies
Regulatory Evolution
Building on the RDEP foundation, regulatory frameworks will continue evolving:
International harmonization: Aligning ultra-rare disease pathways across regions
Flexible evidence requirements: Further accommodating novel trial designs
Real-world data integration: Expanding acceptance of post-marketing evidence
Patient voice amplification: Incorporating patient perspectives throughout development and review
Scientific Innovation
Emerging technologies promise to expand the range of treatable ultra-rare diseases:
Base editing and prime editing: Next-generation gene correction approaches
mRNA therapeutics: Applying COVID-19 vaccine technology to rare genetic diseases
Protein degradation therapies: Novel mechanisms for targeting previously "undruggable" proteins
Cell therapy advances: Expanding beyond cancer to metabolic and neurological conditions
Conclusion
The ultra-rare disease pipeline represents one of the most compelling opportunities in modern pharmaceutical development. With supportive regulatory frameworks like the FDA's RDEP initiative, scientific breakthroughs in genomics and gene therapy, and substantial unmet patient needs, this sector is poised for continued expansion.
Nearly 30 million Americans have a rare disease, but only a few hundred of these conditions currently have FDA approved treatments. This stark reality underscores both the humanitarian imperative and commercial opportunity in ultra-rare disease drug development.
For organizations positioned to navigate the complexities of ultra-rare disease drug development, the potential rewards extend far beyond financial returns. These efforts represent genuine opportunities to transform lives, address previously untreatable conditions, and advance the frontiers of medical science. As the FDA continues to refine its regulatory pathways and scientific understanding deepens, the companies, investors, and institutions that master the unique dynamics of ultra-rare disease pipelines will unlock both extraordinary commercial success and profound humanitarian impact.
The question is no longer whether ultra-rare disease drug development is viable—it's which organizations will lead this therapeutic revolution and capture the billion-dollar potential it represents.
Frequently Asked Questions (FAQs)
Q1: What defines an ultra-rare disease, and how does it differ from a rare disease?
A: According to the FDA, a rare disease affects fewer than 200,000 people in the United States. Ultra-rare diseases typically affect even smaller populations—fewer than 20,000 individuals, with some impacting only hundreds of patients globally. The distinction is important because ultra-rare diseases often require even more innovative clinical trial designs and regulatory approaches due to extremely limited patient populations.
Q2: Why are pharmaceutical companies increasingly interested in ultra-rare disease drug development despite small patient populations?
A: Several factors drive this interest including regulatory advantages from orphan drug designation (seven-year market exclusivity, tax credits, fee waivers), accelerated approval pathways with smaller trial requirements, premium pricing potential due to high unmet need, opportunity for label expansion to broader indications, and strong acquisition interest from larger pharmaceutical companies seeking to diversify their portfolios.
Q3: What is the FDA's Rare Disease Evidence Principles (RDEP) framework, and why is it significant?
A: Announced in September 2025, RDEP provides a flexible regulatory framework specifically designed for ultra-rare genetic diseases with very small patient populations. It allows single pivotal trials, innovative endpoint strategies, and enhanced regulatory interaction to accelerate development timelines while maintaining safety standards. This represents a fundamental shift in how ultra-rare disease treatments can be evaluated and approved.
Q4: How many rare diseases currently have FDA-approved treatments?
A: According to FDA data, there are thousands of rare diseases affecting 25-30 million Americans, but only a few hundred currently have approved treatments. Since the 1983 Orphan Drug Act, more than 600 orphan drug indications have been approved—a dramatic increase from only ten approved treatments before the law's passage.
Q5: What are the biggest challenges in developing treatments for ultra-rare diseases?
A: Key challenges include patient identification and recruitment across multiple countries, limited natural history data to inform endpoint selection, difficulty demonstrating statistical significance with small sample sizes, high manufacturing costs for complex therapies, market access and reimbursement negotiations, and post-marketing safety monitoring with limited patient exposure during development.
Q6: How long does it typically take to develop an ultra-rare disease treatment?
A: Development timelines vary significantly based on therapeutic modality, disease characteristics, and regulatory pathway. With RDEP and accelerated approval mechanisms, some programs can reach approval in 3-5 years from IND filing. However, gene therapies and novel mechanisms may require 7-10 years or longer, particularly if long-term efficacy and safety data are necessary for full approval conversion.
Q7: What role do patient advocacy groups play in ultra-rare disease drug development?
A: Patient advocacy organizations are critical partners, providing patient recruitment support and registry maintenance, natural history data collection, funding for early-stage research, expertise in patient-relevant endpoints, regulatory engagement and patient voice representation, and post-approval real-world evidence generation. Many successful ultra-rare disease programs were catalyzed by patient advocacy efforts.
Q8: What benefits does orphan drug designation provide?
A: According to FDA regulations, orphan drug designation provides seven-year market exclusivity upon approval, tax credits for qualified clinical testing expenses, waiver of FDA application fees, eligibility for orphan drug grant programs, and enhanced access to FDA guidance and support throughout development. These benefits significantly reduce the financial burden of rare disease drug development.
Q9: Are gene therapies the future of ultra-rare disease treatment?
A: Gene therapies represent a transformative approach for genetic ultra-rare diseases, offering potential one-time curative treatments. However, they're one tool among many. The FDA's 2024 approvals included diverse modalities including small molecules, biologics, and enzyme replacement therapies. The optimal approach depends on disease mechanism, and the future likely involves a diverse therapeutic toolkit matched to specific disease characteristics.
Q10: How does the FDA support rare disease drug development?
A: The FDA supports rare disease development through multiple mechanisms including the Office of Orphan Products Development (OOPD), the Rare Disease Innovation Hub launched in 2024, the Rare Disease Evidence Principles (RDEP) framework for ultra-rare genetic diseases, orphan drug designation benefits, grant programs for clinical trials and natural history studies, and early engagement opportunities for developers to receive regulatory guidance throughout the development process.
References
U.S. Food and Drug Administration. (2025). FDA Advances Rare Disease Drug Development with New Evidence Principles.
U.S. Government Accountability Office. (2025). Rare Disease Drugs: FDA Has Steps Underway to Strengthen Coordination of Activities Supporting Drug Development. GAO-25-106774.
U.S. Food and Drug Administration. (2017). FDA's Orphan Drug Modernization Plan.
U.S. Food and Drug Administration. (2024). Novel Drug Approvals for 2024.
U.S. Food and Drug Administration. (2023). Novel Drug Approvals for 2023.
National Institutes of Health. (2025). Rare Diseases.
National Center for Biotechnology Information. (2010). Profile of Rare Diseases - Rare Diseases and Orphan Products. NCBI Bookshelf.
U.S. Food and Drug Administration. Office of Orphan Products Development. Search Orphan Drug Designations and Approvals.
U.S. Food and Drug Administration. Designating an Orphan Product: Drugs and Biological Products.
U.S. Food and Drug Administration. CBER Rare Disease Program.
National Institutes of Health. Genetic and Rare Diseases Information Center (GARD).
U.S. Food and Drug Administration. (2024). CDER Brings Many Safe and Effective Therapies to Patients and Consumers in 2024.
U.S. Food and Drug Administration. (2024). Advancing Health Through Innovation: New Drug Therapy Approvals 2024.
National Institutes of Health Catalyst. NIH Rare Disease Day.
U.S. Food and Drug Administration. NDA and BLA Calendar Year Approvals.
Navigation
© 2026 FyreIgnis Market Research. All rights reserved.
Legal
info@fyreignismarketresearch.com
India
